Congenital malformations can lead to embryonic mortality in many species, and sea turtles are no exception. Genetic and/or environmental alterations occur during early development in the embryo, and may produce aberrant phenotypes, many of which are incompatible with life. Causes of malformations are multifactorial; genetic factors may include mutations, chromosomal aberrations, and inbreeding effects, whereas non-genetic factors may include nutrition, hyperthermia, low moisture, radiation, and contamination. It is possible to monitor and control some of these factors (such as temperature and humidity) in nesting beaches, and toxic compounds in feeding areas, which can be transferred to the embryo through their lipophilic properties.
- congenital malformations
- sea turtle embryos
- epigenetic mechanisms
- environmental factors
- embryonic mortality
1. Introduction
Teratology, from the Greek teras, meaning monster, is the study of causes and mechanisms leading to abnormal development. Developmental malformations are multifactorial, and may include autosomal genetic diseases, point mutations, chromosome aberrations, as well as non-genetic/environmental factors, such as maternal health and nutrition, mechanical problems, exposure to toxicants, radiation or hyperthermia [1]. Teratogens may interfere with developmental processes resulting in congenital malformations; teratogenic agents may act at any time during development, and depending on the severity, the embryo may die or be malformed (Table 1).
Table 1. Summary of embryonic development of sea turtles (stages, processes, and main morphological traits) according to [2][3][4] and prevalent affectations in sea turtles due to teratogenic agents. Images from Miller et al. [4] with the permission of the author and the publisher (CCB, Allen Press Publishing Services); scale bar = 1 mm for stages 7–20; 5 mm for stage 25; 10 mm for stages 26–31; arrows indicate key characteristics.
| Developmental Stage | Developmental Process | Morphological Traits | Prevalent Affectations in Sea Turtles Due to Teratogenic Agents |
|---|---|---|---|
1–9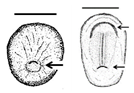 |
Fertilisation-Gastrulation-Early Neurulation After fertilisation, the zygote undergoes cleavage to form a blastocyst; the blastula is reorganised into a trilaminar structure. Neurulation initiates |
Primitive streak; trilaminar embryonic disc composed of endoderm, ectoderm, and mesoderm; notochordal plate; neural groove; neural folds; head fold | The egg is rich in lipid proteins and is ovoposited in middle gastrula stage; depending on the exposure to a teratogenic factors (such as extreme temperatures), embryonic development may be halted [2][3][4] |
10–14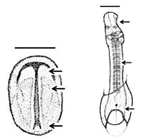 |
Neurulation-Somitogenesis The neural plate folds and transforms into the neural tube; somites form from the paraxial mesoderm of the neurulating embryo |
Neural tube; somites (up to 17); amnion covers half of the embryo; germ layers; amniotic cap; otic vesicle; first pharyngeal cleft; development of the head, brain, heart, and blood vessels | Gnathoschisis (cleft palate) is the prevalent craniofacial malformation in green, loggerhead and olive ridley sea turtles [5][6] caused by an incorrect fusion of the medial nasal and maxillary prominences and frontonsal mass deriving from mesenchymal and neural crest cells [7] |
15–19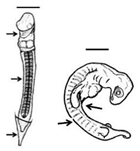 |
Somitogenesis-Organogenesis Organised and integrated processes by which embryonic layers transform into tissues and organs |
Torsion initiates and completes; embryo on left side; somites (>40); amnion, chorion and yolk sac complete; pharyngeal clefts open and start to close; limb buds develop to form digital plates; blood islands visible; tail elongates; lens visible in the eye | Dysmelia (limb differences) is the most common malformation reported for hawksbill turtles [5]; deviations in the signal centre of the apical ectodermal ridge (AER) cause truncated limb bones [8][9] |
20–25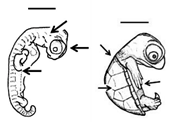 |
Early growth All major organs and systems continue to develop and grow |
All pharyngeal clefts closed; iris pigmented along posterior border; limbs develop; ribs are visible; carapace develops with scutes; tail equals hindlimbs in length, organs and systems continue to grow | Variation in scute patterns is the most common malformation in hard-shelled sea turtles, and is compatible with life [2][3][5]; hot temperatures and dry conditions may produce scute variations [10][11] |
26–31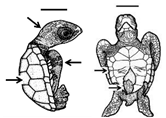 |
Late growth-Hatching Organs and systems conclude growth and development; a fully developed sea turtle is ready to hatch |
Head and flipper scales present; diameter of yolk decreases (piping stage <10 mm); all organs and systems fully developed; pigmentation is evident; hatching occurs | Leucism (hypopigmentation) has been reported for several species of turtles [5][12][13][14][15][16]; these individuals can survive to adulthood, and remain reproductively active [17] |
Embryonic development in sea turtles largely depends on the external environment, and extreme environmental conditions can cause congenital malformations. Sea turtles are ectothermic animals displaying sexual reproduction with internal fertilisation. After mating, females can store viable sperm to fertilise more eggs; fertilization occurs in the upper oviduct, and although no specialized sperm storage structures are present in sea turtles, sperm is stored in the ducts of albumin glands in the upper region of the oviduct [18][19][20]; therefore the offspring of a single female may be from different males [21][22]. Fertilised eggs contain not only all the nutrients necessary for development to occur, but also accumulate xenobiotics, which can be maternally transferred because of their lipophilic properties [23]. Sea turtles search for a suitable place to nest, at which time embryonic development is arrested in middle gastrula stage; once the eggs are laid in the sand, development resumes, which will depend entirely on environmental factors, such as temperature and humidity, until hatchling emergence [21][24][25][2][3][4].
2. Pigmentation Disorders
Leucism (leuc- is the Latin variant of leuk- from the Greek leukos meaning “white”) is a congenital condition showing discoloration of certain parts of the body (hypopigmentation) related to disorders in pigment cell differentiation and/or migration during development producing chromatic aberrations [26]. In leucism, all pigment-producing cells differentiate from the same precursor cell, and therefore it causes a reduction in all types of pigments. Contrary to albinism, in which one or several genes that produce or distribute melanin are defective (showing a complete lack of pigmentation including eyes, skin, and nails), in leucism normal coloration is observed in some parts of the body (such as eyes and nails) [27][28]. Leucism has commonly been referred to as partial albinism, and has been associated with several non-mutually exclusive factors such as pollution [26][29], diet [27], follicular damage [30], genetic mutations [26], or age and sex [26][28]. The lack of pigmentation has also been associated with inbreeding, in which recessive alleles would be expressed [31], indicating low levels of genetic diversity [27]. A leucistic or albino condition may affect fitness, since coloration is an important component for survival (i.e., predator detection) and reproduction (i.e., mate selection) [29].
Leucism has been observed in sea turtle embryos at later stages (sometimes together with other malformations) (Figure 1) [5][32], and they can even reach adulthood [17]. Leucism, when not accompanied by other malformations, can be compatible with life, since the first pigments appear during early growth [3] and total pigmentation appears during late growth when the main characteristic is exponential growth in mass [24], until hatching (Table 1). A possible aetiology could be related to low quality maternal nutrition or exposure to chemical compounds present in the food chain or the water, altering (either by mutational or epigenetic mechanisms) the expression of pigment-producing genes. More than 150 pigmentation-related genes have been identified, harbouring different types of mutations such as single-nucleotide polymorphisms, insertions, deletions, or duplications [32].
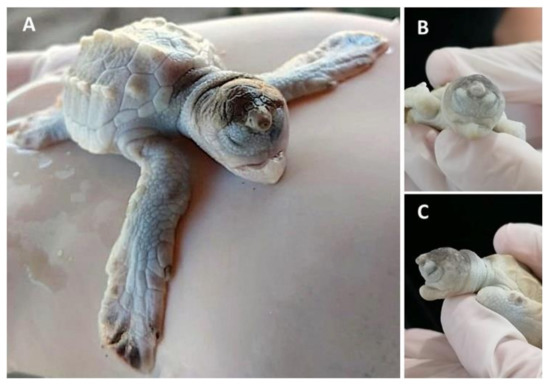
Figure 1. Lepidochelys olivacea embryo with frontal proboscis, orbital hypotelorism (decrease in the distance between the two eyes), leucism (lack of pigmentation), and maxillar agnathia (lack of upper jaw). (A) Whole body view; (B) frontal view; (C) side view. Photographs provided by María Fernanda Calderón-Campuzano.
Interestingly, multiple leucism/albinism has been reported in the loggerhead sea turtle Caretta caretta (22.4% of the nest) [13][15] and the yellow-spotted Amazon River turtle Podocnemis unifilis (43.24% of the nest) [15], suggesting inbreeding as a potential cause, since leucistic individuals were commonly found in small and isolated populations; this isolation could be caused by human activities, leading to a reduction in gene flow [15]. Recently, Madeira et al. [16] reported a clutch of 30 out of 123 (24.4%) albino green sea turtle hatchlings in Poilão Island, Bijagós Archipelago, Guinea-Bissau. The authors reported a normally pigmented female as a mother, but no genetic information about paternity was recorded to know if these organisms originate from the same father. Noteworthy, the body mass and size of leucistic or albino hatchlings were greater than their normal siblings [14][15][16], and this could be an advantage to compensate for the lack of pigments, since these individuals can survive to adulthood, remain reproductively active, and contribute to the population [17]. More studies are needed to understand these differences in size and weight.
3. Craniofacial Disorders
According to previous reports concerning green, loggerhead, and olive ridley sea turtles, craniofacial malformations are the most prevalent [5][6][33]. Craniofacial and brain development are complex interconnected processes in which cephalic neural crest cells are the most important contributors of tissues and sensorial organs [34]. Important signalling regulators for craniofacial development include Sonic hedgehog (Shh) and Wnt, plus families of growth factors (such as fibroblast growth factor (FGF), transforming growth factor (TGF), or epidermic growth factor (EGF)) and retinoic acid (RA), which regulate the interaction of cephalic neural crest cells with other cells; thus gene mutations, chromosomal aberrations, and/or environmental factors affecting these signalling pathways may result in congenital malformations [35].
3.1. Head
The head is a complex structure derived from endoderm, mesoderm, ectoderm, and cephalic neural crest cells. The morphogenetic process involves hundreds of genes interacting to regulate cell proliferation, migration, and differentiation in a coordinated fashion. These regulatory processes are highly dynamic and susceptible to dysregulation [36], illustrated by the large amount of cranial malformations documented in animals, some of which have been observed in sea turtles.
Bicephaly refers to the presence of two heads in a single individual, and has been documented in sea turtles (Figure 2) [5]. According to Velo-Antón et al. [37], the term is used to describe a broad spectrum of developmental alterations, such as the duplication of head structures or incomplete splitting of the zygote related to the occurrence of conjoined twins (which have also been documented in sea turtles, Figure 3). In addition, bicephaly may occur by terminal bifurcation of the notochord during neurulation [36]. Sea turtles may hatch with this condition but do not live long (hours or days) and the causes are unk[1]nown, presumably genetic, environmental or a combination of both [38]. Interestingly, bicephaly is by far the most reported malformation in snakes, both wild and in captivity, and its associated causes could be both cool and hot temperatures during incubation, inbreeding depression, hybridization, environmental pollution, and chemical toxins [38].

Figure 2. Sea turtle embryos with bicephaly (the presence of two heads). (A) Lepidochelys olivacea; (B) Chelonia mydas. Photographs provided by María Fernanda Calderón-Campuzano (A) and Julieta Álvarez-Servín (B).

Figure 3. Conjoined twins (Lepidochelys olivacea). Photograph provided by María Fernanda Calderón-Campuzano.
Microcephaly has also been observed in sea turtle embryos [5] and, according to Pirozzi et al. [39], this is a congenital malformation associated with deficient brain growth, which is determined by the orchestration of tightly regulated processes involving cell proliferation, migration, and death. Mutations (deletions or duplications) in several human genes, defective DNA-repair pathways, as well as chromosome aberrations have been linked to these critical processes impacting brain development [40]. Nevertheless, non-genetic factors, such as environmental conditions, viral infections, and exposure to toxic compounds, may also result in a reduction in brain size [39]. For instance, in humans, specific genetic mutations and some viral infections (i.e., Zika) have been linked to microcephaly [40][41]; however, to our knowledge, no aetiological agents associated with microcephaly have been documented in sea turtles. Because developmental processes are usually conserved, it is possible that similar genetic and non-genetic factors disrupting brain development in humans could also affect brain growth in sea turtles.
Neural tube defects have also been observed in sea turtle embryos [5]; these defects are also multifactorial [42]. Anencephaly occurs during embryogenesis when the neural tube fails to close completely; this condition results from a failed closure of the neural folds in the mid and hindbrain, leading first to exencephaly (also observed in sea turtles), in which the neuroepithelium is exposed, bulging from the brain. This tissue further degenerates, resulting in anencephaly [42]. Encephalocele is another neural tube defect documented in sea turtle embryos; it refers to sac-like protrusions of the brain and its membranes (meninges) caused by failure of the neural tube to close completely. In humans, the genetic basis of neural tube defects is well described, identifying folic acid deficiency as a major causative agent [42][43][44], suggesting nutritional causes. Other non-genetic factors causing neural tube defects such as infections, drugs, caffeine, smoking, and alcohol have been identified in humans, as well as hyperthermia [43]; the latter is relevant in sea turtles since the incubation temperature is crucial during development, and sustained high temperatures may cause congenital malformations. Nevertheless, the effects of hyperthermia on neural tube defects are still inconclusive, since one study did not find evidence that high temperature during sea turtle development causes neural tube defects [10].
3.2. Forebrain and Midline Facial Structures
The most severe phenotypes in the midline facial structure (such as cyclopia, arhinia, and agnathia) identified in sea turtles occur during early development, and could be intrinsically related to the absence or incomplete division of the forebrain into two cerebral hemispheres (i.e., holoprosencephaly), which can also result in mild malformations [45]. Cyclopia has been reported in the olive ridley [5][6]; it is the most severe ocular malformation characterized by the presence of a single eye in the middle of the face due to the presence of a single optic vesicle, and it is always accompanied by multiple craniofacial malformations caused by holoprosencephaly [46]. Arhinia is the absence of nares, and it has been reported in the green turtle [5]. A leatherback sea turtle embryo with holoprosencephaly, arhinia, proboscis, and maxillary micrognathia has been reported, and according to the authors, these malformations could be related to the nutritional status of the mother and/or exposure to mercury [47]. Agnathia (Figure 1) is the absence of jaws (maxilla and/or mandible); it has been identified (although in low proportions) in green, hawksbill, and olive ridley sea turtle embryos [5][6], and it is closely linked to holoprosencephaly [48].
Shh signalling plays a major role in forebrain division, and a loss-of-function of this pathway leads to severe aberrant phenotypes in the mid-craniofacial region [46]. Shh, together with other cell signalling networks (i.e., bone morphogenetic protein (BMP), FGF, Nodal, and RA), controls morphogenetic events for this process and facial development [49]. The proteins of the Hedgehog (HH) family must undergo two post-translational modifications so that they can be secreted as active signalling molecules, first by a cholesterol residue at their N-terminal end, and then by a palmitate residue at their C-terminal end [50][51]. A blockage of Shh signalling in the brain would inhibit the subsequent induction of Shh expression in the frontonasal ectodermal zone (FEZ), a signalling centre that regulates facial development [52]. It is possible that the Shh signalling pathway largely depends on the nutritional status of the mother, with effects on both brain and face development. A deficiency of Shh (i.e., haploinsufficiency) could interact with other genetic and/or epigenetic factors, leading to holoprosencephaly [53], producing aberrant phenotypes with varying degrees of severity. Interestingly, lower blood cholesterol levels have been reported for green turtles with fibropapilloma (as well as with other pathologies) compared to healthy turtles [54][55][56]. Blood cholesterol has been recently proposed as a biomarker of fibropapillomatosis in green turtles, since cholesterol values are significantly lower when the pathology is more severe [57].
RA signalling converges with Shh signalling [58] and coordinates morphogenetic events during brain and face development. RA is an active derivative of vitamin A and is provided exclusively through the diet. The biogenesis of all-trans RA is the first developmental step in the initiation of RA-regulated signalling pathways to subsequently bind to nuclear RA receptors and act as transcription factors [59]. Excess or deficiency of vitamin A is detrimental to normal development [60][61]. The transforming growth factor-beta-induced factor (TGIF) is key in the regulation of RA signalling and is essential to correctly model the forebrain; it regulates the expression of genes controlling both RA synthesis and degradation [62]. Eggs of sea turtles (and of reptiles in general) have a retinoid storage mechanism to prevent hypovitaminosis A [63]. Coincidentally, lower levels of retinol have been reported in diseased green turtles compared to healthy turtles [55][64]. Furthermore, retinoid levels may vary between colonies or populations; in green turtles, significantly different concentrations of vitamin A in plasma have been reported between sampling sites, which was attributed to the availability of food [64]; in loggerhead turtles, it has been reported that plasma concentrations of vitamin A decreased as the nesting season progressed [64], which could indicate maternal transfer to eggs.
Hypoxia-ischemia has also been associated with holoprosencephaly with severe malformations in the midface (cyclopia, arhinia, and micrognathia) [65]. This is relevant in sea turtles since they depend on the exchange of oxygen through the chorion and its availability within the nest. When the forebrain separates to organise the facial structure, the cells of the neural crest begin to migrate, shortly after the closure of the neural tube [66]. This is a population of migratory cells derived from the superficial and neural ectoderm that will give rise to the primordia of the facial skeleton [67]; these cells must undergo an epithelial to mesenchymal transition (EMT) and subsequently migrate to their destinations; FoxD3 and Sox10 are necessary to initiate this process [68], while Sox9 is required for the determination of the chondrogenic lineage [69]. Coincidentally, RA inhibits Sox9 expression [70], while hypoxia induces its expression [71].
Studies in sea turtles indicate that the jaw is the most affected craniofacial region [5][6][33], and in green, loggerhead, and olive ridley turtle embryos, gnathoschisis (cleft palate) is the predominant malformation [5][33]. The FEZ regulates maxillary growth; Shh is expressed in the ectoderm that will form the roof of the mouth and forms a boundary with cells expressing Fgf8 and Wnt9b in the dorsal ectoderm, promoting growth and patterning of the maxilla [72]. Gnathoschisis has its origin in early embryonic development (Table 1) and is caused by an incorrect fusion of the medial nasal and maxillary prominences and frontonsal mass deriving from mesenchymal and neural crest cells [7]. Endochondral bones of the face fail to develop, resulting in multiple defects such as gnathoschisis or brachygnathia, which may be promoted by Sox9 dysregulation [73]. Brachygnathia is characterized by shortening of the jaw (maxillary or mandibular) and has been identified in the green sea turtle as recurrent as gnathoschisis [5].
It appears that the spatial organization of gene expression patterns in FEZ is highly associated with the facial shape [74], and changes in the expression domains of signalling molecules such as Shh, Fgf8, Wnt9b, Bmp2, Bmp4, and Bmp7 could result in a variety of aberrant phenotypes of the jaw and the nasal region in sea turtle embryos. For instance, prognathia and agnathia, which refer to the lengthening and absence, respectively, of the jaw, maxillary or mandibular, have been recurrently identified in olive ridley turtle embryos (Figure 1) [5][6]; laterognathia (characterized by lower jaw turned to right or left) has been reported in live hawksbill embryos, although at low frequencies [5]; and prosoposchisis (a fissure of the face extending from the jaw to the eye) has also been reported in olive ridley turtle embryos [5].
3.3. Eyes
Eye development begins with the division of the ocular field into two optic vesicles [46]. The Pax6 gene, as part of a set of genes that encode the highly conserved ocular field transcription factor among vertebrates [75], is considered an eye selector gene necessary to initiate the regulatory cascade for lens and eye development [76]. Hence, a loss-of function on Pax6 can cause anophthalmia (Figure 4A) [77]. After cyclopia, it is the most severe phenotype characterized by absence of the eyes. In green turtle embryos, anophthalmia has been reported as prevalent, above gnathoschisis [5].

Figure 4. Lepidochelys olivacea embryos with (A) right anophthalmia (absence of the right eye,) and (B) amelia (absence of flipper) and schistosomus reflexus syndrome (ventral view). Photographs taken from supplementary file in [5]with the permission of the authors and publisher (John Wiley & Sons).
Pax6 together with Rax, Six3 and Lhx2 constitute a group of eye field transcription factors (EFTFs) necessary for ocular development [75]. Sox2 and Otx2 activate the expression of Rax, inducing up-regulation of the EFTFs. In olive ridley sea turtle embryos, microphthalmia (abnormally small eyes) has been reported, although in very low frequencies [5]. In humans, Sox2 and Otx2 are both the major causative factors for anophthalmia and severe microphthalmia [78].
The RA signalling pathway could be involved in ocular malformations, since severe ocular abnormalities, including microphthalmia, have been observed in mice whose mothers were treated with RA [79]. Paradoxically, a loss of function of the Stra6 gene, a cellular receptor that binds to the retinol-binding protein and plays an important role in the transport of retinol to the eye [80], is also a cause of microphthalmia and anophthalmia in humans [81]. These reports suggest a nutritional aetiology; however, in addition to nutritional factors, pollutants and gas exchange in the embryo may contribute to craniofacial malformations. Variations in temperature and humidity can also generate aberrant phenotypes [3][82][83], although the mechanisms of action are not totally understood.
Macrophthalmia, characterized by abnormally big eyes, has also been reported in olive ridley embryos [5]. In humans and mice, haploinsufficiency of CRIM1 (cysteine-rich transmembrane bone morphogenetic protein (BMP) regulator 1) has been reported as the cause of macrophthalmia [84]. Interestingly, contaminants such as polycyclic aromatic hydrocarbons (PAHs) can induce transcriptional changes on CRIM1 and disrupt multiple cellular processes [85]. Synophthalmia, also reported for olive ridley turtle embryos [5], is characterized by eyes more or less completely fused into one, and is related to the incomplete division of the optic field.
3.4. Nares
From the FEZ, two nasal placodes emerge, which are then invaded by growing ectoderm and mesenchyme, and then fuse to become the nasal cavity and the primitive choana. The expression of Fgf8 is required for proper morphogenesis of the entire nasal cavity [86], and recently, SMCHD1 (structural maintenance of chromosomes flexible hinge-domain containing protein 1) has emerged as a key regulator of embryonic genomic function as an epigenetic modifier in several genes [87]. In humans, mutations in the SMCHD1 gene have been identified as causing arhinia with different degrees of severity, and Bosma Arhinia Microphthalmia Syndrome [88].
In sea turtle embryos, malformations in the nares are less common compared to malformations of the jaw and eyes [5][6][33]. Rinoschisis, a peculiar and rare malformation, characterized by vertical separation of the nares into two equal parts, has been identified in hawksbill turtle embryos [5]; in humans, mutations in the FREM1 gene (FRAS1-related extracellular matrix protein 1) have been reported to cause this malformation [89]. Another rare malformation is rhinocephaly, consisting of one proboscis-shaped nare generally in the front of the head; it has been reported for olive ridley and leatherback turtle embryos [5][47] related to holoprosencephaly.
This entry is adapted from the peer-reviewed paper 10.3390/ani11020444
References
- Ujházy, E.; Mach, M.; Navarová, J.; Brucknerová, I.; Dubovický, M. Teratology—Past, present and future. Interdiscipl. Toxicol. 2012, 5, 163–168, doi:10.2478/v10102-012-0027-0.
- Kaska, Y.; Downie, R. Embryological development of sea turtles (Cheloniamydas, Carettacaretta) in the Mediterranean. Zool. Middle East 1999, 19, 55–69, doi:10.1080/09397140.1999.10637796.
- Miller, J.D. Embryology of marine turtles. In Biology of the Reptilia; Gans, C., Northcutt, R.G., Ulinsky, P., Eds.; Academic Press: New York, NY, USA, 1985; pp. 269–328.
- Miller, J.D.; Mortimer, J.A.; Limpus, C.J. A Field Key to the Developmental Stages of Marine Turtles (Cheloniidae) with Notes on the Development of Dermochelys. Chelonian Conserv. Biol. 2017, 16, 111–122, doi:10.2744/CCB-1261.1.
- Bárcenas-Ibarra, A.; De la Cueva, H.; Rojas-Lleonart, I.; Abreu-Grobois, F.A.; Lozano-Guzmán, R.I.; Cuevas, E.; García-Gasca, A. First approximation to congenital malformation rates in embryos and hatchlings of sea turtles. Birth Defects Res. Part A 2015, 103, 203–224, doi:10.1002/bdra.23342.
- Bárcenas-Ibarra, A.; Maldonado-Gasca, A. Malformaciones en Embriones y Neonatos de Tortuga Golfina (Lepidochelys olivacea) en Nuevo Vallarta, Nayarit, México. Vet. Mexico 2009, 40, 371–380. Available online: http://www.scielo.org.mx/pdf/vetmex/v40n4/v40n4a3.pdf (accessed on December 20, 2020).
- Abramyan, J.; Richman, J.M. Recent insights into the morphological diversity in the amniote primary and secondary palates. Dev. Dyn. 2015, 244, 1457–1468, doi:10.1002/dvdy.24338.
- Towers, M.; Tickle, C. Growing models of vertebrate limb development. Development 2009, 136, 179–190, doi:10.1242/dev.024158.
- Pownall, M.E.; Isaacs, H.V. FGF Signalling in Vertebrate Development; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010; p. 75, doi:10.4199/C00011ED1V01Y201004DEB002.
- Zimm, R.; Bentley, B.P.; Wyneken, J.; Moustakas-Verho, J.E. Environmental causation of turtle scute anomalies in ovo and in silico. Integr. Comp. Biol. 2017, 57, 1303–1311, doi:10.1093/icb/icx066.
- Telemeco, R.S.; Warner, D.A.; Reida, M.K.; Janzen, F.J. Extreme developmental temperatures result in morphological abnormalities in painted turtles (Chrysemys picta): A climate change perspective. Integr. Zool. 2013, 8, 197–208, doi:10.1111/1749-4877.12019.
- Craven, K.S.; Sheppard, S.; Stallard, L.B.; Richardson, M.; Belcher, C.N. Investigating a Link Between Head Malformations and Lack of Pigmentation in Loggerhead sea Turtle Embryos (Caretta caretta) in the Southeastern United States. Herpetology Notes 2019, 12, 819–825. Available online: https://www.biotaxa.org/hn/article/viewFile/45607/50090 (accessed on December 20, 2020).
- Marcovaldi, M.; Paes-e-Lima, E.; Penteado, R. Albino sea Turtles Hatchlings in Brazil. Mar. Turt. Newsl. 1995, 69, 10. Available online: http://www.seaturtle.org/mtn/archives/mtn69/mtn69p10a.shtml (accessed on December 20, 2020).
- Godfrey, M.H.; Mrosovsky, N. Comment on albino sea turtle hatchlings in Brazil. Mar. Turt. Newsl. 1995, 69, 10–11. Available online: http://www.seaturtle.org/mtn/archives/mtn69/mtn69p10b.shtml (accessed on December 20, 2020).
- Erickson, J.; Kaefer, I.L. Multiple Leucism in a Nest of the Yellow-Spotted Amazon River Turtle, Podocnemis unifilis. Salamandra 2014, 51, 273–276. Available online: http://www.salamandra-journal.com (accessed on December 20, 2020).
- Madeira, F.M.; Patrício, A.R.; Indjai, B.; Barbosa, C.; Regalla, A.; Catry, P.; Rebelo, R. High number of healthy albino green turtles from Africa’s largest population. Mar. Turt. Newsl. 2020, 160, 19. Available online: http://www.seaturtle.org/mtn/archives/mtn160/mtn160-5.shtml (accessed on December 20, 2020).
- Restrepo, J.; Valverde, R.A. Leucistic Adult Female Green Sea Turtles (Chelonia mydas) Successfully Nesting at Tortuguero, Costa Rica. Mar. Turt. Newsl. 2019, 159, 23–25. Available online: http://www.seaturtle.org/mtn/archives/mtn159/mtn159-5.shtml (accessed on January 23, 2021).
- Gist, D.H.; Jones, J.M. Sperm storage within the oviduct of turtles. J. Morphol. 1989, 199, 379–384, doi:10.1002/jmor.1051990311.
- Rostal, D.C.; Robeck, T.R.; Owens, D.W.; Kraemer, D.C. Ultrasound imaging of ovaries and eggs in Kemp’s Ridley sea turtles (Lepidochelys kempi). J. Zoo Wildl. Med. 1990, 21, 27–35. Available online: http://www.jstor.org/stable/20095016 (accessed on February 1, 2021).
- Rostal, D.C. Reproductive physiology of the ridley sea turtle. In Biology and Conservation of Ridley Sea Turtles; Plotkin, P.T., Ed.; Johns Hopkins University Press: Baltimore, MD, USA, 2007; pp. 151–165.
- Spotila, J.R. Sea Turtles: A Complete Guide to Their Biology, Behavior, and Conservation; Johns Hopkins University Press: Baltimore, MD, USA, 2004; p. 240.
- Jensen, M.P.; Abreu-Grobois, F.A.; Frydenberg, J.; Loeschcke, V. Microsatellites provide insight into contrasting mating patterns in arribada vs. non-arribada olive ridley sea turtle rookeries. Mol. Ecol. 2006, 15, 2567–2575, doi:10.1111/j.1365-294X.2006.02951.x.
- Van de Merwe, J.P.; Hodge, M.; Whittier, J.M.; Ibrahim, K.; Lee, S.Y. Persistent organic pollutants in the green sea turtle Chelonia mydas: Nesting population variation, maternal transfer, and effects on development. Mar. Ecol. Prog. Ser. 2010, 403, 269–278, doi:10.3354/meps08462.
- Andrews, R. Patterns of embryonic development. In Reptilian Incubation: Environment, Evolution and Behaviour; Deeming, C., Ed.; Nottingham University Press: Nottingham, UK, 2004; pp. 75–102.
- Plotkin, P. Nomadic behaviour of the highly migratory olive ridley sea turtle Lepidochelys olivacea in the eastern tropical Pacific Ocean. Endanger Species Res. 2010, 13, 33–40, doi:10.3354/esr00314.
- Van Grouw, H. What colour is that bird? Br. Birds 2013, 106, 17–29.
- Brito, J.; Valdivieso-Bermeo, K. First records of leucism in eight species of small mammals (Mammalia: Rodentia). Therya 2016, 7, 483–489, doi:10.12933/therya-16-408.
- Izquierdo, L.; Thomson, RL.; Aguirre, JI.; Díez-Fernández, A.; Faivre, B.; Figuerola, J.; Ibáñez-Álamo, D. Factors associated with leucism in the common blackbird Turdus merula. J. Avian Biol. 2018, E01778, doi: 10.1111/jav.01778.
- Møller, A.P.; Mousseau, T.A. Mutation and sexual selection: A test using barn swallows from Chernobyl. Evolution 2003, 57, 2139–2146, doi:10.1554/03-051.
- Phillips, A.R. The Cause of Partial Albinism in a Great-tailed Grackle. Wilson Bull. 1954, 66, 66. Available online: https://sora.unm.edu/sites/default/files/journals/wilson/v066n01/p0066-p0066.pdf (accessed on December 20, 2020).
- Bensch, S.; Hansson, B.; Hasselquist, D.; Nielsen, B. Partial albinism in a semi-isolated population of Great Reed Warblers. Hereditas 2000, 133, 167–170, doi:10.1111/j.1601-5223.2000.t01-1-00167.x.
- Cieslak, M.; Reissmann, M.; Hofreiter, M.; Ludwig, A. Colours of domestication. Biol. Rev. 2011, 86, 885–899, doi: 10.1111/j.1469-185X.2011.00177.x.
- Drenen, J.D. Occurrence of Physical Abnormalities in Caretta caretta at Hobe Sound National Wildlife Refuge, 1987 and 1988. Mar. Turt. Newsl. 1990, 48, 19–20. Available online: http://www.seaturtle.org/mtn/archives/mtn48/mtn48p19.shtml (accessed on December 20, 2020).
- Creuzet, S.E.; Martinez, S.; Le Douarin, N.M. The cephalic neural crest exerts a critical effect on forebrain and midbrain development. Proc. Natl. Acad. Sci. USA 2006, 103, 14033–14038, doi:10.1073/pnas.0605899103.
- Ornoy, A. Craniofacial malformations and their association with brain development: The importance of a multidisciplinary approach for treatment. Odontology 2020, 108, 1–15, doi:10.1007/s10266-019-00433-7.
- Twigg, S.R.F.; Wilkie, A.O.M. New insights into craniofacial malformations. Hum. Mol. Genet. 2015, 24, R50–R59, doi:10.1093/hmg/ddv228.
- Velo-Antón, G.; Buckley, D.; Daoudi, A.D.; Cordero-Rivera, A. Bicephaly in Salamandra salamandra larvae. Herpetol. Bull. 2007, 101, 31–33.
- Wallach, V. Axial Bifurcation and Duplication in Snakes: Part 1: A Synopsis of Authentic and Anecdotal Cases; Maryland Herpetological Society: Annapolis, Maryland, USA, 2007; Volume 43, p. 57.
- Pirozzi, F.; Nelson, B.; Mirzaa, G. From Microcephaly to Megalencephaly: Determinants of Brain Size. Clinical Research. Dialogues Clin. Neurosci. 2018, 20, 267–283. Available online: www.dialogues-cns.org (accessed on December 20, 2020).
- Gilmore, E.C.; Walsh, C.A. Genetic causes of microcephaly and lessons for neuronal development. WIREs Dev. Biol. 2013, 2, 461–478, doi:10.1002/wdev.89.
- Wang, J-N.; Ling, F. Zika virus infection and microcephaly: Evidence for a causal link. Int. J. Environ. Res. Public Health 2016, 13, 1031, doi:10.3390/ijerph13101031.
- Greene, N.D.E.; Copp, A.J. Neural tube defects. Annu. Rev. Neurosci. 2014, 37, 221–242, doi:10.1146/annurev-neuro-062012-170354.
- Detrait, E.R.; George, T.M.; Etchevers, H.C.; Gilbert, J.R.; Vekemans, M.; Speer, M.C. Human neural tube defects: Developmental biology, epidemiology, and genetics. Neurotoxicol. Teratol. 2005, 27, 515–524, doi:10.1016/j.ntt.2004.12.007.
- Bassuk, A.G.; Kibar, Z. Genetic Basis of Neural Tube Defects. Semin Pediatr. Neurol. 2009, 16, 101–110, doi:10.1016/j.spen.2009.06.001.
- Shiota, K.; Yamada, S. Early pathogenesis of holoprosencephaly. Am. J. Med. Genet. Part C Semin. Med. Genet. 2010, 154, 22–28, doi:10.1002/ajmg.c.30248.
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 1996, 383, 407–413, doi:10.1038/383407a0.
- Choi, E.; Charles, K.E.; Charles, K.L.; Stewart, K.M.; Morrall, C.E.; Dennis, M.M. Leatherback sea turtle (Dermochelys coriacea) embryo and hatchling pathology in Grenada, with comparison to St. Kitts. Chelonian Conserv. Biol. 2020, 19, 111–123, doi:10.2744/CCB-1395.1.
- Dennis, J.F.; Kurosaka, H.; Iulianella, A.; Pace, J.; Thomas, N.; Beckham, S.; Williams, T.; Trainor, P.A. Mutations in Hedgehog acyltransferase (Hhat) perturb Hedgehog signaling, resulting in severe acrania-holoprosencephaly-agnathia craniofacial defects. PLoS Genet. 2012, 8, e1002927, doi:10.1371/journal.pgen.1002927.
- Petryk, A.; Graf, D.; Marcucio, R. Holoprosencephaly: Signaling interactions between the brain and the face, the environment and the genes, and the phenotypic variability in animal models and humans. WIRES Dev. Biol. 2015, 4, 17–32, doi:10.1002/wdev.161.
- Cooper, M.K.; Wassif, C.A.; Krakowiak, P.A.; Taipale, J.; Gong, R.; Kelley, R.I.; Porter, F.D.; Beachy, P.A. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat. Genet. 2003, 33, 508–513, doi:10.1038/ng1134.
- Buglino, J.A.; Resh, M.D. Hhat is a palmitoylacyltransferase with specificity for N‐palmitoylation of Sonic Hedgehog. J. Biol. Chem. 2008, 283, 22076–22088, doi:10.1074/jbc.M803901200.
- Marcucio, R.S.; Cordero, D.R.; Hu, D.; Helms, J.A. Molecular interactions coordinating the development of the forebrain and face. Dev. Biol. 2005, 284, 48–61, doi:10.1016/j.ydbio.2005.04.030.
- Kietzman, H.W.; Everson, J.L.; Sulik, K.K.; Lipinski, R.J. The teratogenic effects of prenatal ethanol exposure are exacerbated by Sonic Hedgehog or GLI2 haploinsufficiency in the mouse. PLoS ONE 2014, 9, e89448, doi:10.1371/journal.pone.0089448.
- Aguirre, A.A.; Balazs, G.H. Blood biochemistry values of green turtles, Chelonia mydas, with and without fibropapillomatosis. Comp. Haematol. Int. 2000, 10, 132–137, doi:10.1007/s005800070004.
- Bloodgood, J.C.; Norton, T.M.; Hoopes, L.A.; Stacy, N.I.; Hernandez, S.M. Comparison of hematological, plasma biochemical, and nutritional analytes of rehabilitating and apparently healthy free-ranging Atlantic green turtles (Chelonia mydas). J. Zoo Wildl. Med. 2019, 50, 69–81, doi:10.1638/2017-0250.
- De Mello, D.M.; Alvarez, M.C. Health assessment of juvenile green turtles in southern São Paulo State, Brazil: A hematologic approach. J. Vet. Diagn. Invest. 2020, 32, 25–35, doi:10.1177/1040638719891972.
- Da Silva, C.C.; Bianchini, A. Blood cholesterol as a biomarker of fibropapillomatosis in green turtles. Mar. Turt. Newsl. 2019, 158, 16–21, doi:10.1016/j.aquatox.2015.11.007.
- Helms, J.A.; Kim, C.H.; Hu, D.; Minkoff, R.; Thaller, C.; Eichele, G. Sonic hedgehog participates in craniofacial morphogenesis and is down-regulated by teratogenic doses of retinoic acid. Dev. Biol. 1997, 187, 25–35, doi:10.1006/dbio.1997.8589.
- Ghyselinck, N.B.; Duester, G. Retinoic acid signaling pathways. Development 2019, 146, dev167502, doi:10.1242/dev.167502.
- Sucov, H.M.; Evans, R.M. Retinoic acid and retinoic acid receptors in development. Mol. Neurobiol. 1995, 10, 169–184, doi:10.1007/BF02740674.
- McCaffery, P.J.; Adams, J.; Maden, M.; Rosa‐Molinar, E. Too much of a good thing: Retinoic acid as an endogenous regulator of neural differentiation and exogenous teratogen. Eur. J. Neurosci. 2003, 18, 457–472, doi:10.1046/j.1460-9568.2003.02765.x.
- Gongal, P.A.; Waskiewicz, A.J. Zebrafish model of holoprosencephaly demonstrates a key role for TGIF in regulating retinoic acid metabolism. Hum. Mol. Genet. 2008, 17, 525–538, doi:10.1093/hmg/ddm328.
- Irie, T.; Sugimoto, T.; Ueki, N.; Senoo, H.; Seki, T. Retinoid storage in the egg of reptiles and birds. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2010, 157, 113–118, doi:10.1016/j.cbpb.2010.05.009.
- Frutchey, K.P. Plasma Levels of Vitamins A and E in Marine Turtles (CheIonia mydas and Caretta caretta). Master’s Thesis, University of Central Florida, Orlando, FL, USA, 2004. Available online: https://stars.library.ucf.edu/cgi/viewcontent.cgi?article=5579&context=rtd (accessed on January 23, 2021).
- Siebert, J.R. Cyclopia, aprosencephaly, and acardiac twinning: Is hypoxia‐ischemia a unifying mechanism? Am. J. Med. Genet. Part A 2007, 143, 3100–3106, doi:10.1002/ajmg.a.32027.
- Hou, L.; Takeuchi, T. Neural crest development in reptilian embryos, studied with monoclonal antibody, HNK-1. Zool. Sci. 1994, 11, 423–423.
- Le Dréau, G.; Martí, E. Dorsal–ventral patterning of the neural tube: A tale of three signals. Dev. Neurobiol. 2012, 72, 1471–1481, doi:10.1002/dneu.22015.
- Betancur, P.; Bronner-Fraser, M.; Sauka-Spengler, T. Assembling neural crest regulatory circuits into a gene regulatory network. Annu. Rev. Cell Dev. Biol. 2010, 26, 581–603, doi:10.1146/annurev.cellbio.042308.113245.
- Mori-Akiyama, Y.; Akiyama, H.; Rowitch, D.H.; De Crombrugghe, B. Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc. Natl. Acad. Sci. USA 2003, 100, 9360–9365, doi:10.1073/pnas.1631288100.
- Sekiya, I.; Koopman, P.; Tsuji, K.; Mertin, S.; Harley, V.; Yamada, Y.; Shinomiya, K.; Niguji, A.; Noda, M. Transcriptional suppression of Sox9 expression in chondrocytes by retinoic acid. J. Cell. Biochem. 2001, 81, 71–78, doi:10.1002/jcb.1077.
- Amarilio, R.; Viukov, S.V.; Sharir, A.; Eshkar-Oren, I.; Johnson, R.S.; Zelzer, E. HIF1α regulation of Sox9 is necessary to maintain differentiation of hypoxic prechondrogenic cells during early skeletogenesis. Development 2007, 134, 3917–3928, doi:10.1242/dev.008441.
- Hu, D.; Marcucio, R.S.; Helms, J.A. A zone of frontonasal ectoderm regulates patterning and growth in the face. Development 2003, 130, 1749–1758, doi:10.1242/dev.00397.
- Lee, Y.H.; Saint‐Jeannet, J.P. Sox9 function in craniofacial development and disease. Genesis 2011, 49, 200–208, doi:10.1002/dvg.20717.
- Young, N.M.; Hu, D.; Lainoff, A.J.; Smith, F.J.; Diaz, R.; Tucker, A.S.; Trainor, P.A.; Schneider, R.A.; Hallgrimsson, B.; Marcucio, R.S. Embryonic bauplans and the developmental origins of facial diversity and constraint. Development 2014, 141, 1059–1063, doi:10.1242/dev.099994.
- Zaghloul, N.A.; Yan, B.; Moody, S.A. Step‐wise specification of retinal stem cells during normal embryogenesis. Biol. Cell 2005, 97, 321–337, doi:10.1042/BC20040521.
- Kozmik, Z. Pax genes in eye development and evolution. Curr. Opin. Genet. Dev. 2005, 15, 430–438, doi:10.1016/j.gde.2005.05.001.
- Glaser, T.; Jepeal, L.; Edwards, J.G.; Young, S.R.; Favor, J.; Maas, R.L. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat. Genet. 1994, 7, 463–471, doi:10.1038/ng0894-463.
- Gerth-Kahlert, C.; Williamson, K.; Ansari, M.; Rainger, J.K.; Hingst, V.; Zimmermann, T.; Tech, S.; Guthoff, R.F.; Van Heyningen, V.; FitzPatrick, D.R. Clinical and mutation analysis of 51 probands with anophthalmia and/or severe microphthalmia from a single center. Mol. Genet. Genomic Med. 2013, 1, 15–31, doi:10.1002/mgg3.2.
- Sulik, K.K.; Dehart, D.B.; Rogers, J.M.; Chernoff, N. Teratogenicity of low doses of all‐trans retinoic acid in presomite mouse embryos. Teratology 1995, 51, 398–403, doi:10.1002/tera.1420510605.
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825, doi:10.1126/science.1136244.
- Williamson, K.A.; FitzPatrick, D.R. The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur. J. Med. Genet. 2014, 57, 369–380, doi:10.1016/j.ejmg.2014.05.002.
- Lynn, W.G.; Ullrich, M.C. Experimental production of shell abnormalities in turtles. Copeia 1950, 1950, 253–262, doi:10.2307/1437903.
- Burger, J. Effects of incubation temperature on hatchling pine snakes: Implications for survival. Behav. Ecol. Sociobiol. 1998, 43, 11–18, doi:10.1007/s002650050461.
- Beleggia, F.; Li, Y.; Fan, J.; Elcioğlu, N.H.; Toker, E.; Wieland, T.; Maumenee, I.H.; Akarsu, N.A.; Meitinger, T.; Strom, M.; et al. CRIM1 haploinsufficiency causes defects in eye development in human and mouse. Hum. Mol. Genet. 2015, 24, 2267–2273, doi:10.1093/hmg/ddu744.
- Lu, X.; Shao, J.; Li, H.; Yu, Y. Early whole-genome transcriptional response induced by benzo [a] pyrene diol epoxide in a normal human cell line. Genomics 2009, 93, 332–342, doi:10.1016/j.ygeno.2008.12.007.
- Kawauchi, S.; Shou, J.; Santos, R.; Hébert, J.M.; McConnell, S.K.; Mason, I.; Calof, A.L. Fgf8 expression defines a morphogenetic center required for olfactory neurogenesis and nasal cavity development in the mouse. Development 2005, 132, 5211–5223, doi:10.1242/dev.02143.
- Schall, P.Z.; Ruebel, M.L.; Latham, K.E. A new role for SMCHD1 in life’s master switch and beyond. Trends Genet 2019, 35, 948–955, doi:10.1016/j.tig.2019.10.001.
- Shaw, N.D.; Brand, H.; Kupchinsky, Z.A.; Bengani, H.; Plummer, L.; Jones, T.I.; Samocha, K. SMCHD1 mutations associated with a rare muscular dystrophy can also cause isolated arhinia and Bosma arhinia microphthalmia syndrome. Nat. Genet. 2017, 49, 238, doi:10.1038/ng.3743.
- Alazami, A.M.; Shaheen, R.; Alzahrani, F.; Snape, K.; Saggar, A.; Brinkmann, B.; Bavi, P.; Al-Gazali, L.I.; Alkuraya, F.S. FREM1 mutations cause bifid nose, renal agenesis, and anorectal malformations syndrome. Am. J. Hum. Genet. 2009, 85, 414–418, doi:10.1016/j.ajhg.2009.08.010.