Protease-activated receptors (PARs) are a class of G protein-coupled receptors (GPCRs) with a unique mechanism of activation, prompted by a proteolytic cleavage in their N-terminal domain that uncovers a tethered ligand, which binds and stimulates the same receptor.
- protease-activated receptors
- serine proteases
- G protein-coupled receptors
- matrix metalloproteinases
1. Protease-activated receptors (PARs): activation and signaling
Protease-activated receptors (PARs) are a family of G protein-coupled receptors (GPCRs), which includes four members (PAR1, PAR2, PAR3, and PAR4) belonging to group A rhodopsin-like GPCR subfamily. PARs have an exclusive mechanism of activation, which requires a site-specific proteolytic cleavage in their N-terminal extracellular domain. This exposes a tethered ligand that binds to the same receptor, activating it [1].
Prototypical PARs activators are serine proteases, firstly recognized as coagulation factors, like thrombin, tissue plasminogen activator (tPA), factor Xa (FXa), factor VIIa (FVIIa), activated protein C (APC), and plasmin. Other PARs activators are trypsins, proteases released from leukocytes, like cathepsin G, elastase, and proteinase 3, as well as cell-surface proteases as membrane-type serine protease 1 (MT-SP1), and the cysteine protease, calpain [1][2][3]. Additionally, PARs can be activated by various matrix metalloproteinases (MMPs), such as MMP-1, MMP-2, MMP-3, MMP-8, MMP-9, and MMP-13, by proteolysis at non-canonical sites [3][4]. While some proteases can activate multiple PARs, other ones specifically cleave one PAR subtype (Table 1). Actually, thrombin activates PAR1, PAR3, and PAR4, although with different potencies, but does not stimulate PAR2, which is instead cleaved by trypsin and tryptase, besides other coagulation factors. The same protease can produce opposite effects on different PARs subtypes, as in the case of cathepsin G, a neutrophil serine protease, that disarms PAR1, by cleaving it into non-functional parts, while activating PAR2 and PAR4, by proteolysis that release tethered ligands [1][2][3][4][5].
Table 1. Protease-activated receptors (PARs): activation, brain localization, and signaling.
| Receptor | Activating Proteases |
Inactivating Proteases |
Activating Peptides |
Signaling Pathways | Cerebral Localization |
|---|---|---|---|---|---|
| PAR1 | Thrombin Factor VIIa (FVIIa) Factor Xa (FXa) Plasmin MMP-1, -2, -3, -8, -9, -12, -13 Activated protein C (APC) Elastase Proteinase 3 Kallikrein 4, -5, -6, -14 Granzyme A, B, K Calpain-1 |
Cathepsin G Proteinase 3 Elastase Plasmin Chymase |
SFLLR-NH2 TFLLR-NH2 NPNDKYEPF-NH2 PRSFLLR-NH2 |
Gq Gi G12/13 β-arrestins |
Hippocampus Cortex Amydgala Substantia nigra Ventral tegmental Area Thalamus Hypothalamus Striatum Dorsal root ganglion |
| PAR2 | Trypsin I/II Trypsin IV Tryptase Factor VIIa (FVIIa) Factor Xa (FXa) Elastase Proteinase 3 Cathepsin G Acrosin Granzyme A Kallikrein 2, -4,-5, -6, 14 |
Chymase | SLIGRL-NH2 SLIGKV-NH2 AC-98170 AC-55541 |
Gq Gi G12/13 β-arrestins |
Hippocampus Cortex Amydgala Substantia nigra Thalamus Hypothalamus Striatum Dorsal root ganglion |
| PAR3 | Thrombin Trypsin Activated protein C (APC) |
Cathepsin G | TFRGAP-NH2 | Gq | Hippocampus Cortex Amydgala Thalamus Hypothalamus Striatum Dorsal root ganglion |
| PAR4 | Thrombin Trypsin Plasmin Cathepsin G MT-SP1 |
GYPGQV-NH2 GYPGKF-NH2 AYPGKF |
Gq G12/13 |
Hippocampus Cortes Amydgala Thalamus Hypothalamus |
In addition to the proteolytic activation, PARs can be stimulated by short peptides corresponding with the tethered ligand sequence. These peptides are able to induce PARs stimulation in the absence of proteolytic cleavage because they replace endogenous PARs-bound ligands in the activation-binding sites. Such alternative modality of activation allows a more controlled PARs activation, and is useful for distinguishing PARs functions devoid of side-effects due to protease-dependent cleavage of additional targets. The PAR1-tethered ligand peptide is SFLLR-NH2. Besides PAR1, it also activates PAR2, though with a minor efficacy [6], but modification in the first amino acid leads to TFLLR-NH2, which is a specific PAR1 activator. The PAR2-tethered sequence is SLIGKV-NH2, while TFRGAP-NH2 is the tethered ligand for PAR3, and GYPGQV-NH2 for PAR4 [1] (Table 1).
PARs activation elicits an intricate network of intracellular signaling pathways. PARs are coupled to various G proteins—Gq, Gi, and G12/13—and, additionally, they can also activate G protein-independent signaling mechanisms [1][2][3][4][5][6][7]. More information is available on PAR1, which is the first member of PARs’ family to be identified. Canonical PAR1 activation, consequent to cleavage-induced exposition of SFLLR-NH2 sequence, results in multiple G protein-dependent and G protein-independent signaling pathways (Figure 1). Through Gαq, PAR1 activates phospholipase C β (PLCβ), thereby triggering phosphoinositide hydrolysis, with the generation of inositol-1,4,5-trisphosphate (IP3) and diacyl-glycerol (DAG), thus, leading to Ca2+ mobilization from intracellular stores and activation of protein kinase C (PKC). This results in the activation of various Ca2+-regulated kinases and phosphatase. PAR1 coupling with Gα12/13, that binds to guanine nucleotide exchange factors (GEFs), results in the activation of the small soluble G protein, Rho, and consequently of Rho-activated kinases. Furthermore, PARs activation, by coupling with Gi/o, induces the inhibition of adenylyl cyclase (AC), whereas via the βγ subunit, induces the opening of K+ channels (namely G protein-activated inward rectifying K+ channels, GIRK), the activation of G protein-coupled receptor kinases (GRKs), as well as the stimulation of non-receptor tyrosine kinases, and the activation of phosphotydil-inositole-3-kinase (PI3K), that induces the activation of other kinases signaling pathways, including mitogen-activated protein kinase (MAPK) [1][2][4][7] (Figure 1).
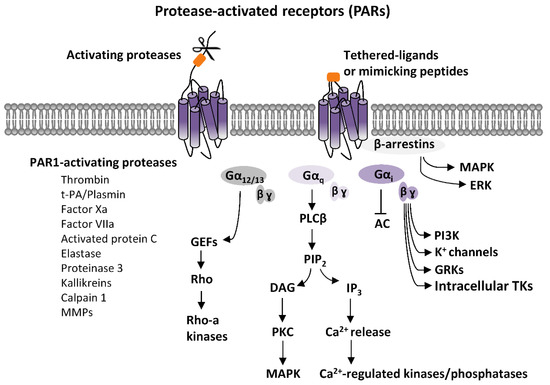
Figure 1. Protease-activated receptors (PARs) signaling. Scheme of the principal PARs-dependent signaling pathways. PARs activation, elicited by proteases-induced unmasking of tethered ligands or, alternatively, by ligand-mimicking peptides, stimulates various G proteins-dependent pathways. Gαq-mediated activation of phospholipase C β (PLCβ) results in the hydrolysis of phosphatidylinositol and generation of inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG), that fosters Ca2+ intracellular mobilization from internal stores, and activation of Ca2+-regulated kinases and phosphatase. Protein kinase C (PKC), which is also activated by DAG, stimulates mitogen-activated protein kinases (MAPK). PARs coupling with Gα12/13, that binds to guanine nucleotide exchange factors (GEFs), results in the activation of the small soluble G protein, Rho, and, consequently, of Rho-activated kinases. PARs-induced activation of Gi, inhibits, via Gαi,, adenylyl cyclase (AC), and, via the βγ subunit, produces opening of G protein-activated inward rectifying K+ channels (GIRK), the activation of G protein-coupled receptor kinases (GRKs), as well as the stimulation of intracellular tyrosine kinases (TKs), and the activation of phosphotydil-inositole-3-kinase (PI3K), that then activates other kinases signaling pathways, including mitogen-activated protein kinase (MAPK). Additionally, PARs stimulations can activate G protein-independent mechanisms, mediated by the recruitment of β-arrestins and activation of diverse signaling pathways, including MAPK-like extracellular signal-regulated kinases (ERK).
2. PARs activation in the Brain
PARs have well-recognized roles in coagulation, hemostasis, and inflammation, and have been deeply investigated for their function in cellular survival/degeneration processes [1]. In addition to effects in peripheral systems, it is becoming overt that PARs have important roles in the central nervous system (CNS). Research of such functions has been alimented by early detections of cerebral expression of different PARs subtypes [8][9][10][11][12][13]. PARs’ family includes four members (PAR1, PAR2, PAR3, and PAR4). PAR1, which is the first to be identified, has been originally termed as‘thrombin receptor’ [14]. Beyond its initial identification in platelets, PAR1 expression has been reported in several organs and different cell types, including endothelial cells, fibroblasts, monocytes, T-cell lines, smooth muscle cells, and in organs such as stomach, colon, kidney, testis, eye, and brain [1]. In the brain, PAR1 is ubiquitously expressed, being found in the prefrontal cortex, basal ganglia, dorsal striatum, nucleus accumbens, substantia nigra, ventral tegmental area, amygdala, and hippocampus. Its localization has been reported either in neurons, or in astrocytes and microglia, even though there are strong differences among different brain areas and cellular populations [8][10][11][15][16][17]. PAR2 has been identified following PAR1 as a receptor for the serine protease trypsin [18]. PAR2 expression has been reported in both the human and rodent CNS in various areas, including the hippocampus (through CA1, CA2, and CA3 areas and the granular cell layer of the dentate gyrus), as well as cortex, amygdala, thalamus, hypothalamus, substantia nigra, and striatum [9][11][19][20]. PAR3, earlier described as a second thrombin receptor [21], displays a similar cerebral expression of PAR2, being localized in various hippocampal and cortical areas, as well as in amygdala, thalamus, hypothalamus, and striatum [11]. Brain localization of PAR4, firstly known as a receptor for both thrombin and trypsin, has been described in the hippocampus and cortex, thalamus, hypothalamus, and amygdala [11]. While it is documented that all PARs display a broad expression in the various brain areas, and some indication on regions of highest expression have been revealed, a deeper analysis to expose overlapping vs. segregate expression of distinct subtypes in sub-regions or cellular populations remains to be completed.
Evidence of brain expression of PARs has been complemented with the demonstration of resident sources of PARs-activating proteases in the brain. Such data has challenged the earlier belief about PARs activation in the brain occurring only in pathological conditions allowing influx of peripheral proteases, through an impaired blood-brain barrier (BBB), and it is now accepted that PARs-activating proteases can be released from neurons, astrocytes, microglia, or other immunity cells that are resident (or recruited) in the brain, in addition to being derived from the circulation [1][11][14]. Specifically, there is evidence documenting brain synthesis of the prototypical PARs activator, thrombin, with both a mature or precursor form, pro-thrombin, found in several brain areas [8][13][15][22][23][24]. Likewise, tPA can be released by neurons, glial cells, and endothelial cells, being highly expressed in various brain regions, including the cerebellum, cortex, amygdala, and hippocampus [24][25][26][27][28][29]. Further evidence supports brain expression of other PARs-activating proteases, including trypsin [30], and trypsin-like proteases, such as MSP and kallikreins [31][32], besides MMPs [33]. Actions of cerebral PARs-activating proteases is tightly regulated. Serine proteases activity in CNS is tempered by another class of proteins, i.e., the serine protease inhibitors (SERPINs), including protease nexin-1 (PN-1), neuroserpin, and antithrombin 3 (AT3) [24][34][35][36]. The activity of such SERPINs, by influencing PARs-activating proteases, can indirectly affect PARs signaling/function in the brain.
Hence, it is currently established that, in a normal brain, there are necessary elements—PARs activators and receptors—to permit physiological PARs signaling. Beyond such a physiological tone, levels of PARs-activating proteases possibly boost during some conditions, like inflammation or trauma, that either recruit additional proteases-releasing cells types, or increase BBB permeability, fostering coagulation cascade proteases inflow in the CNS from the periphery [1], with the consequence of an abnormal PARs activation. Actually, multifaceted PARs roles have been previously reported in neuroinflammatory and neurodegenerative processes in diverse cerebral illnesses, in stroke, brain trauma, Alzheimer’s disease (AD), and Parkinson’s disease (PD). Likewise, aberrant activity of serine proteases and MMPs, possibly resulting in abnormal PARs signaling, has been linked to AD, PD, TBI, stroke, epilepsy, and familial encephalopathy with neuroserpin inclusion bodies (FENIB) [3][5][33][37][38][39][40][41][42]. Differently from their pathological relevance, physiological roles for PARs in the brain have been less appreciated. Nevertheless, it is becoming clear that PARs have a “neuromodulatory” function, affecting neurotransmission and synaptic plasticity in a normal brain, thus, possibly contributing to either learning and memory processes and complex behaviors.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22020869
References
- Ossovskaya, V.S.; Bunnett, N.W. Protease-activated receptors: Contribution to physiology and disease. Physiol. Rev. 2004, 84, 579–621.
- Gieseler, F.; Ungefroren, H.; Settmacher, U.; Hollenberg, M.D.; Kaufmann, R. Proteinase-activated receptors (PARs)—Focus on receptor-receptor-interactions and their physiological and pathophysiological impact. Cell Commun. Signal. 2013, 11, 1–26.
- Heuberger, D.M.; Schuepbach, R.A. Protease-activated receptors (PARs): Mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. J. Thromb. 2019, 17, 1–24.
- Fox, O.W.; Preston, R.J.S. Molecular basis of protease-activated receptor 1 signaling diversity. J. Thromb. Haemost. 2020, 18, 6–164.
- Ramachandran, R.; Noorbakhsh, F.; DeFea, K.; Hollenberg, M.D. Targeting proteinase-activated receptors: Therapeutic potential and challenges. Nat. Rev. Drug Discov. 2012, 11, 69–86.
- Blackhart, B.D.; Emilsson, K.; Nguyen, D.; Teng, W.; Martelli, A.J.; Nystedt, S.; Sundelin, J.; Scarborough, R.M. Ligand cross-reactivity within the protease-activated receptor family. J. Biol. Chem. 1996, 271, 16466–16471.
- Soh, U.J.; Dores, M.R.; Chen, B.; Trejo, J. Signal transduction by protease-activated receptors. Br. J. Pharm. 2010, 160, 191–203.
- Weinstein, J.R.; Gold, S.J.; Cunningham, D.D.; Gall, C.M. Cellular localization of thrombin receptor mRNA in rat brain: Expression by mesencephalic dopaminergic neurons and codistribution with prothrombin mRNA. J. Neurosci. 1995, 15, 2906–2919.
- D’Andrea, M.R.; Derian, C.K.; Leturcq, D.; Baker, S.M.; Brunmark, A.; Ling, P.; Darrow, A.L.; Santulli, R.J.; Brass, L.F.; Andrade-Gordon, P. Characterization of protease-activated receptor-2 immunoreactivity in normal human tissues. J. Histochem. Cytochem. 1998, 46, 157–164.
- Niclou, S.P.; Suidan, H.S.; Pavlik, A.; Vejsada, R.; Monard, D. Changes in the expression of protease-activated receptor 1 and protease nexin-1 mRNA during rat nervous system development and after nerve lesion. Eur. J. Neurosci. 1998, 10, 1590–1607.
- Striggow, F.; Riek-Burchardt, M.; Kiesel, A.; Schmidt, W.; Henrich-Noack, P.; Breder, J.; Krug, M.; Reymann, K.G.; Reiser, G. Four different types of protease-activated receptors are widely expressed in the brain and are up-regulated in hippocampus by severe ischemia. Eur. J. Neurosci. 2001, 14, 595–608.
- Wang, H.; Ubl, J.J.; Reiser, G. Four subtypes of protease-activated receptors, co-expressed in rat astrocytes, evoke different physiological signaling. Glia 2002, 37, 53–63.
- Sokolova, E.; Reiser, G. Prothrombin/thrombin and the thrombin receptors PAR-1 and PAR-4 in the brain: Localization, expression and participation in neurodegenerative diseases. Thromb. Haemost. 2008, 100, 576–581.
- Vu, T.K.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068.
- Dihanich, M.; Kaser, M.; Reinhard, E.; Cunningham, D.; Monard, D. Prothrombin mRNA is expressed by cells of the nervous system. Neuron 1991, 6, 575–581.
- Balcaitis, S.; Xie, Y.; Weinstein, J.R.; Andersen, H.; Hanisch, U.; Ransom, B.R.; Möller, T. Expression of proteinase-activated receptors in mouse microglial cells. Neuroreport 2003, 14, 2373–2377.
- Junge, C.E.; Lee, C.J.; Hubbard, K.B.; Zhang, Z.; Olson, J.J.; Hepler, J.R.; Brat, D.J.; Traynelis, S.F. Protease-activated receptor-1 in human brain: Localization and functional expression in astrocytes. Exp. Neurol. 2004, 188, 94–103.
- Nystedt, S.; Emilsson, K.; Wahlestedt, C.; Sundelin, J. Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 9208–9212.
- Bushell, T.J.; Plevin, R.; Cobb, S.; Irving, A.J. Characterization of proteinase-activated receptor 2 signalling and expression in rat hippocampal neurons and astrocytes. Neuropharmacology 2006, 50, 714–725.
- Lohman, R.J.; O’Brien, T.J.; Cocks, T.M. Protease-activated receptor-2 regulates trypsin expression in the brain and protects against seizures and epileptogenesis. Neurobiol. Dis. 2008, 30, 84–93.
- Ishihara, H.; Connolly, A.J.; Zeng, D.; Kahn, M.L.; Zheng, Y.W.; Timmons, C.; Tram, T.; Coughlin, S.R. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature 1997, 386, 502–506.
- Turgeon, V.L.; Houenou, L.J. The role of thrombin-like (serine) proteases in the development, plasticity and pathology of the nervous system. Brain Res. Brain Res. Rev. 1997, 25, 85–95.
- Turgeon, V.L.; Salman, N.; Houenou, L.J. Thrombin: A neuronal cell modulator. Thromb Res. 2000, 99, 417–427.
- Almonte, A.G.; Sweatt, J.D. Serine proteases, serine protease inhibitors, and protease-activated receptors: Roles in synaptic function and behavior. Brain Res. 2011, 1407, 107–122.
- Qian, Z.; Gilbert, M.E.; Colicos, M.A.; Kandel, E.R.; Kuhl, D. Tissue plasminogen activator is induced as an immediate-early gene during seizure, kindling, and long-term potentiation. Nature 1993, 361, 453–457.
- Sallés, F.J.; Strickland, S. Localization and regulation of the tissue plasminogen activator-plasmin system in the hippocampus. J. Neurosci. 2002, 22, 2125–2134.
- Shin, C.Y.; Kundel, M.; Wells, D.G. Rapid, activity-induced increase in tissue plasminogen activator is mediated by metabotropic glutamate receptor-dependent mRNA translation. J. Neurosci. 2004, 24, 9425–9433.
- Yepes, M.; Lawrence, D.A. New functions for an old enzyme: Nonhemostatic roles for tissue-type plasminogen activator in the central nervous system. Exp. Biol. Med. 2004, 226, 1097–1104.
- Lochner, J.E.; Honigman, L.S.; Grant, W.F.; Gessford, S.K.; Hansen, A.B.; Silverman, M.A.; Scalettar, B.A. Activity-dependent release of tissue plasminogen activator from the dendritic spines of hippocampal neurons revealed by live-cell imaging. J. Neurobiol. 2006, 66, 564–577.
- Gschwend, T.P.; Krueger, S.R.; Kozlov, S.V.; Wolfer, D.P.; Sonderegger, P. Neurotrypsin, a novel multidomain serine protease expressed in the nervous system. Mol. Cell Neurosci. 1997, 9, 207–219.
- Scarisbrick, I.A.; Isackson, P.J.; Ciric, B.; Windebank, A.J.; Rodriguez, M. MSP, a trypsin-like serine protease, is abundantly expressed in the human nervous system. J. Comp. Neurol. 2001, 431, 347–361.
- Bernett, M.J.; Blaber, S.I.; Scarisbrick, I.A.; Dhanarajan, P.; Thompson, S.M.; Blaber, M. Crystal structure and biochemical characterization of human kallikrein 6 reveals that a trypsin-like kallikrein is expressed in the central nervous system. J. Biol. Chem. 2002, 277, 24562–24570.
- Brkic, M.; Balusu, S.; Libert, C.; Vandenbroucke, R.E. MMPs in brain disease friends or foes: Matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediat. Inflam. 2015, 2015, 1–27.
- Wagner, S.L.; Van Nostrand, W.E.; Lau, A.L.; Farrow, J.S.; Suzuki, M.; Bartus, R.T.; Schuppek, R.; Nguyen, A.; Cotman, C.W.; Cunningham, D.D. Co-distribution of protease nexin-1 and protease nexin-2 in brains of non-human primates. Brain Res. 1993, 626, 90–98.
- Reinhard, E.; Suidan, H.S.; Pavlik, A.; Monard, D. Glia-derived nexin/protease nexin-1 is expressed by a subset of neurons in the rat brain. J. Neurosci Res. 1994, 37, 256–270.
- Miranda, E.; Lomas, D.A. Neuroserpin: A serpin to think about. Cell Mol. Life Sci. 2006, 63, 709–722.
- Junge, C.E.; Sugawara, T.; Mannaioni, G.; Alagarsamy, S.; Conn, P.J.; Brat, D.J.; Chan, P.H.; Traynelis, S.F. The contribution of protease-activated receptor 1 to neuronal damage caused by transient focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2003, 100, 13019–13024.
- Nicole, O.; Goldshmidt, A.; Hamill, C.E.; Sorensen, S.D.; Sastre, A.; Lyuboslavsky, P.; Helper, J.R.; McKeon, R.J.; Traynelis, S.F. Activation of protease-activated receptor-1 triggers astrogliosis after brain injury. J. Neurosci. 2005, 25, 4319–4329.
- Luo, W.; Wang, Y.; Reiser, G. Protease-activated receptors in the brain: Receptor expression, activation, and functions in neurodegeneration and neuroprotection. Brain Res. Rev. 2007, 56, 331–345.
- Davis, R.L.; Shrimpton, A.E.; Holohan, P.D.; Bradshaw, C.; Feiglink, D.; Collins, G.H.; Sonderegger, P.; Kinter, J.; Becker, L.M.; Lacbawan, F.; et al. Familial dementia caused by polymerization of mutant Neuroserpin. Nature 1999, 401, 376–379.
- Molinari, F.; Meskanaite, V.; Munnich, A.; Sonderegger, P.; Colleaux, L. Extracellular proteases and their inhibitors in genetic diseases of the central nervous system. Hum. Mol. Genet. 2003, 12, R195–R200.
- Fabbro, S.; Seeds, N.W. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J. Neurochem. 2009, 109, 303–315.