Adrenocortical carcinoma (ACC) is a heterogenous group of diseases with different clinical be-haviour between adult and paediatric patients. In addition, three histological variants, oncocytic, myxoid and sarcomatoid are noted on the recent World Health Organisation (WHO) classification of ACC. A review of recent literature showed that the different types of ACC have distinctive demographic data, clinical presentation, pathology, biological behaviour, genomic and patients’ prognosis. In addition, recent updates of pathology staging for ACC allow refinement of prognos-tic grouping for planning treatment of the patients with ACC.
- adrenocortical carcinoma
- adrenal
- staging
- pathology
- oncocytic
- myxoid
- sarcomatoid
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Adrenal cortical carcinoma (ACC) is a rare cancer but is the most common primary cancer in the adrenal gland [1]. It is the second most common malignant tumour of the endocrine organ after anaplastic thyroid carcinoma [2]. In large pathology series, ACC accounts for 6.8% of all primary adrenal tumours [3]. In the most recent population database (based on approximately 2000 cases) from the Surveillance, Epidemiology, and End Results (SEER) published in 2018 from the United States of America (USA) of ACC-diagnosed patients between 1973 to 2012, the annual incidence of the cancer was 1.02 per one million [4]. This figure has reflected the increase incidence of the cancer due to increased use of sensitivity imaging studies in current years. In addition, in a recent large series of adrenal incidentalomas (n = 3672), ACC accounts for 1.4% of cases [5].
There is an increase of new knowledge about ACC in terms of genomic characterisation based on the findings of The Cancer Genome Atlas (TCGA) [6–9]. Further genomic studies showed that mutational and expression profiles of advanced and metastatic ACC are very similar to those from primary ACC as well as low mutation rates, few major oncogenic drivers and loss of function mutations in several epigenetic regulators suggest an epigenetic basis of ACC such as DNA methylation [10,11]. In a similar period, the eighth American Joint Committee on Cancer (AJCC) Cancer Staging Manual and UICC (Union for International Cancer Control) Tumour, Lymph node, Metastasis (TNM) staging Manual published in 2016 (adopted for use in 2017) have updated the pathological staging of ACC based on the European Network for the study of Adrenal Tumours (ENSAT) [12]. In addition, the fourth edition of the World Health Organization (WHO) book series on tumour classification published in 2017 has updated the histological classification of ACC [13].
2. Conventional Adrenocortical Carcinoma
2.1. Demographic Characteristics
Based on the small number of cases of other histological variants of ACC, conventional ACC comprises more than 90% of cases. Thus, data noted in epidemiological studies likely represent the features of conventional ACC. Like other endocrine tumours, ACC is more common in females. From the pooled series of different countries, ACC is more common in females with male to female ratio of approximately 1 to 1.4 [3,14,15]. The average age at presentation of patients with ACC in larger series range from 47- to 55-year-olds [15]. Although some series demonstrate a bimodal age distribution with peaks in the paediatric group and middle age group, SEER data [4] show that there is a steady increase of ACC with increased age peaking at the sixth and seventh decades.
2.2. Clinical Features
Approximately half of ACC is functioning with signs and symptoms of hormone (s) secretion [16]. Patients with functioning ACC were younger, more likely to be females and present with metastatic disease. Of functioning tumours, almost half present with signs or symptoms of cortisol excess (Cushing syndrome). The second most common functional presentation was sex hormone secretion accounting for 20% of cases. These sex hormone-producing ACCs are mainly androgens. Rarely, oestrogen secreting/feminising ACC were reported [17–21]. Aldosterone secreting ACC is uncommon and accounted for approximately 8% of cases. In addition, mixed hormone production was seen in 15% to 25% of functioning ACCs [15,22].
Non-functioning ACCs commonly present with abdominal mass, abdominal pain as well as general symptoms of malignancy. Paraneoplastic manifestations of having ACC such as hypoglycaemia (due to insulin growth factor 2 [IGF2] production) [23] and clinical manifestations related to adrenocorticotropic hormone ACTH production have been reported [24]. Rare manifestations of patients with ACC include cancer-related thrombotic microangiopathy [25] and tumour rupture with retroperitoneal haemorrhage [26–28].
ACC could be a cancer in the setting of several hereditary syndromes. These syndromes could occur in high as 5–10% of patients with ACC [29]. In adult patients with ACC, these syndromes include multiple endocrine neoplasia type 1 (MEN1, approximately 20 cases reported) [30], Lynch syndrome (mismatch repair genes) (approximately 10 cases) [31], Li-Fraumeni syndrome (TP53, more than 10 cases) [32] and neurofibromatosis type 1 (NF1, approximately 10 cases) [33]. Rarely, ACC can occur in patients with Carney complex (protein kinase A regulatory subunit 1A [PRKARIA]) [34,35], Gardner’s syndrome [36] and familial adenomatous polyposis (adenomatous polyposis coli [APC]) [37,38]. Although extremely rare, ACCs have been reported in patients with congenital adrenal hyperplasia and myelolipoma [39–41]. It is likely these are collision tumours rather than having been aetiologically related.
2.3. Pathology
ACC is often noted on left adrenal (left to right ratio = 1.2 to 1). Bilateral ACC is uncommon and accounted for approximately 1% of the cases [4]. Ectopic ACCs, presumably arise from ectopic adrenal rests, have been reported in the retroperitoneum [42], ovary [43,44], spinal region [45], liver [46] and abdominal wall [47]. A case of retroperitoneal ACC was noted in 68-year-old man with Lynch syndrome [48].
On macroscopic examination, ACC is often yellow to tan colour (Figure 1). Areas of necrosis and haemorrhage are common which give a heterogenous appearance on cut sections. The tumour is often large with median maximum dimension of 100 mm to 120 mm [4,49]. From the findings in recent literature, the largest non-functioning ACC was reported in a 39-year-old Greek woman with a maximum dimension of 237 mm [50] whereas the largest functioning (androgen-secreting) ACC was noted in a 48-year-old Canadian woman with a maximum dimension of 230 mm [51]. Also, Weiss and colleagues have mentioned an ACC with 280 mm in dimension in their series published in 1989 [52] but no clinicopathological details about the case. The tumour could be seen to grossly extend to adjacent organs as well as to regional veins and the right atrium which could lead to pulmonary embolism [53].
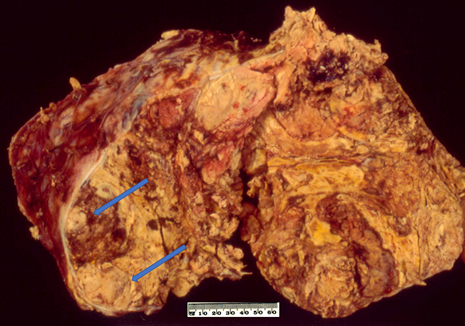
Figure 1. Conventional adrenocortical carcinoma (ACC) showing variegated cut surface of yellow tumour with foci of haemorrhages and necrosis (arrows).
Upon microscopic examination, ACC often has eosinophilic cytoplasm. There are often thick fibrous bands and capsules (Figure 2A). Necrosis is common (Figure 2B). Mitotic figures are often prominent. For prognostic purposes, ACC was subdivided into low grade or high grade depending on mitotic frequency (dividing line is between ≤20 and >20 mitosis per 50 high power fields/10 mm2) [52].
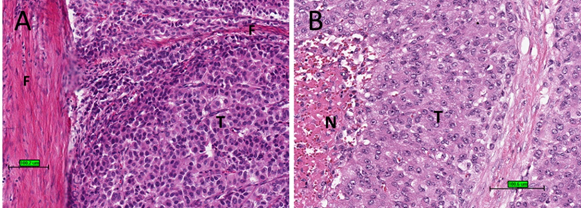
Figure 2. Histological features of ACC. (A) Tumour cells (T) with eosinophilic cytoplasm with thick fibrous bands (F). (B) Tumour cells (T) with necrosis (N) (haematoxylin and eosin stain) (scale bar–100 µm).
It is often not difficult to differentiate ACC from benign cortical adenoma or borderline cortical tumour. Nevertheless, in some instances, the diagnosis of ACC needs to rely on assessment of multiple pathological parameters. The most widely accepted system adopted by WHO classification for assessment is Weiss criteria published in 1984 [52,54]. Using this system, ACC could be diagnosed on at least three of the 9 histological features—high nuclear grade (Fuhrman III or IV), high mitotic rate (>5 mitoses per 50 high power field, atypical mitotic figures, ≤25% clear cells, diffuse architecture, tumour confluent necrosis, venous invasion, sinusoidal invasion, and capsular invasion. Modified Weiss has been proposed which is based on 5 of the 9 histological features of Weiss (mitotic counts, clear cells, atypical mitotic figures, tumour necrosis and capsular invasion) [55]. In this system, mitotic rate and clear cells have twice the points (‘weight”) compared with other features. The outcome of use of modified Weiss is highly correlated with the original Weiss system [55].
Diffuse architecture must be present in >33% and could be identified by disruption of the reticulin [56] and interruption of basal lamina (antibodies to laminin or collagen type IV). A reticulin algorithm has been used for diagnosis of ACC which involves an abnormal/absent reticulum framework and at least one of the 3 of histological features (tumour necrosis, presence of venous invasion and mitotic rate of >5/50 high power field) [56,57].
These systems for predicting malignant potential of adrenocortical tumours are applicable mainly in conventional ACC. It is important to note that no single microscopic criterion on its own is indicative of malignancy and there is subjective variability in the interpretation [58]. Studies have proposed the use of proliferative index (Ki-67 index > 5%) (Figure 3) [59] and IGF2 over-expression to confirm the diagnosis of ACC [60].
A relatively new system published by a European group in 2015 is the Helsinki score which relay on mitotic rate, necrosis and Ki-67 index (3× mitotic count [>5/50 high power fields] + 5× presence of necrosis + Ki-67 proliferative index in the most proliferative index of the tumour) of ACC and focus on the predicting diagnosis as well as prognosis of ACC [61,62]. A Helsinki score >8.5 is associated with metastatic potential and warrant the diagnosis of ACC.
The most common malignant tumour in adrenal gland is metastatic carcinoma [63]. In a biopsy specimen, it is important to differentiate ACC from metastatic carcinoma as well as phaeochromocytoma by clinical history, biochemical studies, radiology, and a panel of immunohistochemical stains [64]. ACC expresses markers specific for steroid-producing cells which often include steroidogenic factor-1 (SF-1) and inhibin alpha (Figure 4). The tumour is also positive for markers expressed by other tumour types, such as melan A and calretinin. In addition, abnormal beta-catenin intracellular accumulation is often noted [15]. Common epithelial markers such as cytokeratin, EMA, CEA are generally negative. Although ACC could be positive for synaptophysin, it is negative for chromogranin.
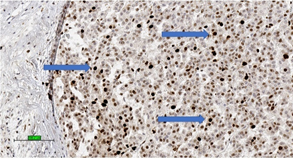
Figure 3. Adrenocortical carcinoma of high Ki-67 index with high percentage of nuclear stain (brown colour) for Ki-67 (3,3′-diaminobenzidine/haematoxylin stains) (blue arrows) (scale bar—100 µm).
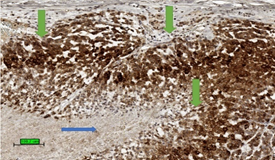
Figure 4. Tumour stain positive (brown colour) for inhibin (3,3′-diaminobenzidine/haematoxylin stains) in cytoplasm of ACC (green arrows). The necrotic area is not stained up (blue arrow). (scale bar—100 µm).
Distant metastases at presentation occurred in approximately one third of the patients with ACC [15]. The common sites of distant metastases of ACC are liver, lung, and bone [65]. Unusual descripted metastatic locations of ACC have been reported in the stomach [66], pancreas [67], skin [68], spleen [69], tongue [70] and brain including meninges [71,72]. Recurrence and distant metastases of ACC often occur quickly (within 2 years). However, late locoregional recurrence could occur 18 years after surgery [73] and liver metastasis have been reported in a patient 23 years after surgical resection of ACC [74].
2.4. Prognosis
Prognosis of the conventional ACC depends on the cancer stage [12]. In addition, the prognostic value of Helsinki score has been demonstrated to be an outperforming prognostic index [56]. In addition, large tumour size and cortisol-secreting tumour were additional factors for ACC-specific death [49]. Moreover, there are many predictive features proposed for the prognosis of patients with ACC such as mitotic grading [52], Ki-67 index [75], mi-RNAs expression [76], expression of PDL-1 [77], SF-1 [78] and sterol-O-acyl transferase 1 (SOAT1) [79]. Preliminary data also shows that for localised ACC, molecular makers (expression, methylation, and chromosome alterations) could predict cancer recurrence [9]. Nevertheless, many of these markers need validation and some (such as molecular markers) are difficult to apply in clinical settings.
Overall, the prognosis of patients with ACC is poor. A recent Danish study on 160 patients with ACC showed that a median survival of patients with ACC was 35 months [14] whereas in the study based on the USA SEER database, the median survival of patients with ACC was 17 months [4]. The 5-year cancer specific survival rate of patients with ACC as noted from USA SEER database was 38% [1]. It is worth noting that surgery on the primary site even in metastatic ACC significantly improved overall and cancer-specific survival of patients with ACC [80]. The 5 year-survival of adult patients from multiple datasets with ACC after surgery range from 40% to 70% [49,81]. The estimated five-year overall survival rate for patients with ACC in recent cohorts is slightly less than 50% [11]
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9020175