Despite recent advances in first-line treatment for small-cell lung cancer (SCLC), durable respons-es remain rare. The DNA repair enzyme poly-(ADP)-ribose polymerase (PARP) was identified as a therapeutic target in SCLC using unbiased preclinical screens and confirmed in human and mouse models. Early trials of PARP inhibitors, either alone or in combination with chemotherapy, showed promising but limited responses, suggesting that selecting patient subsets and treatment combinations will prove critical to further clinical development. Expression of SLFN11 and other components of the DNA damage response (DDR) pathway appears to select for improved re-sponses. Combining PARP inhibitors with agents that damage DNA and inhibit DDR appears particularly effective in preclinical and early trial data, as well as strategies that enhance anti-tumor immunity downstream of DNA damage.
- SCLC
- PARP
- DDR
- ICB
- synthetic lethality
- SLFN11
- STING
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Small-cell lung cancer (SCLC) is a high-grade neuroendocrine malignancy with a poor prognosis that accounts for 13% of all lung cancer diagnoses [1,2]. First-line treatment for extensive-stage SCLC (ES-SCLC) is often effective, with a response rate of more than 60% to platinum-based chemotherapy, but prior to recent first-line advances, median overall survival was less than 11 months [2,3]. Immune checkpoint blockade (ICB) using inhibitors of the programmed cell death protein and its ligand (PD-1/PD-L1) initially showed promise in the third-line setting, and inclusion into first-line platinum-based therapy has demonstrated an overall survival benefit, becoming the new standard of care [4–7] with particular improvement in durable responses. Until recently, topotecan has been the only option approved by the United States Food and Drug Administration (FDA) in the second-line setting but has not been widely utilized due to concerns about toxicity and only modest efficacy [8–10]. Despite this, multiple randomized studies with a topotecan control arm have been negative, highlighting the resistant disease state [11–13] after prior platinum-based therapy. One of the negative studies that failed to meet its primary overall survival endpoint was a recent combination of lurbinectedin and doxorubicin compared to a control arm of either topotecan or CAV (cyclophosphamide, doxorubicin, vincristine) [14], which followed prior accelerated FDA approval of single-agent lurbinectedin based upon impressive data in small-cell lung cancer from a basket trial [15]. National Comprehensive Cancer Network guidelines include multiple regimens that may be considered in the second-line setting and beyond, but clinical trial is one of the three preferred regimens, highlighting the need for more effective treatments [16].
SCLC is a transcriptionally active disease with common (up to 90%) loss-of-function genomic alterations in the tumor suppressor genes TP53 and RB1, creating further genomic instability by preventing arrest of the cell cycle for important DNA repair [17–20]. This suggests the potential for synergy with treatments that disrupt replication enough to halt the process and lead to apoptosis. One such approach, poly-(ADP)-ribose polymerase (PARP) inhibitors, have been a compelling class of drugs in the ongoing efforts to improve outcomes in this cancer that has been so resistant to other treatment options. Overexpression of PARP1 in SCLC further suggests therapeutic potential for PARP inhibitors [21].
Recurrent, targetable genomic alterations have not been identified in SCLC, but epigenetic and gene expression studies have led to the description of four distinct molecular subtypes defined by transcriptional regulators [22]. Subtyping of SCLC may offer an opportunity for better identification of treatment options with a higher likelihood of generating durable responses and will likely be an important component of prospective studies, including those evaluating PARP inhibitors and combinations.
PARP inhibitors represent a therapeutic class that has become an important treatment option for multiple tumor types. Although there is evidence of response, PARP inhibitors are not currently part of the treatment armamentarium for SCLC, and single-agent efficacy is limited. There is substantial ongoing investigation incorporating PARP inhibitors into the treatment of SCLC, and the following sections outline the mechanisms and rationale for these promising therapeutic combinations.
2. PARP Inhibitor Mechanism of Action
Recognition and repair of DNA damage form an essential cellular function mediated by a number of interconnected pathways termed the DNA damage response (DDR; Figure 1). PARP enzymes are a family of proteins that function in recognition and repair of DNA breaks, chromatin remodeling, and transcriptional regulation [23]. PARP 1 and 2 enzymatic function is activated by binding single-strand DNA breaks (SSB) and involves poly-ADP ribosylation (PARylation) of various substrates and recruitment of proteins that mediate DNA repair (Figure 1). PAR groups are subsequently metabolized by Poly-(ADP)-ribose glycohydrolase (PARG) and other enzymes as part of coordinated dePARylation critical to effective DNA repair [23]. In the absence of SSB repair by PARP1, the replication fork stalls and double strand breaks occur prompting repair via homologous recombination (HR) or non-homologous end joining (NHEJ). If DSBs are not correctly repaired, replication aberrancies such as mutations, deletions, chromosomal translocations, and amplifications can occur resulting in cell death, senescence or malignant transformation. PARP inhibitors were initially developed to sensitize tumor cells to standard treatments such as chemotherapy or radiation, which induce DNA damage [24]. However, the observation that tumor cells with defects in HR are highly sensitive to single-agent PARP inhibition accelerated their clinical development [25,26]. The activity of PARP inhibitors in patients with BRCA1 or BRCA2 mutant cancers was the first clinical demonstration of synthetic lethality for cancer therapy [27]. In this setting, by inhibiting PARP catalytic activity and trapping PARP on DNA, PARP inhibitors stall replication machinery leading to DNA double strand breaks (DSB). In the absence of BRCA1 or BRCA2, these breaks cannot be repaired by HR (Figure 1). Several PARP inhibitors are currently approved or in clinical trials. In addition to differences in their selectivity for PARP 1/2, these agents differ in their PARP trapping function, with talazoparib being the most potent [28]. Further studies in tumors without HR deficiency suggest that PARP inhibitors could have a broader role in cancer therapy [29].
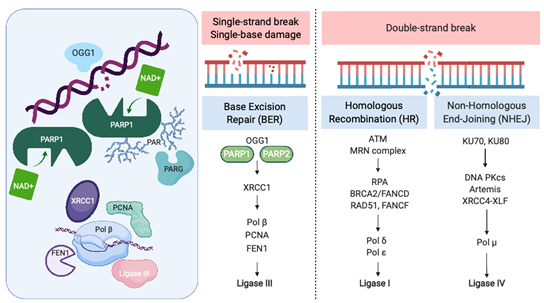
Figure 1. The Role of PARP in the DNA Damage Response. PARP = poly-(ADP)-ribose polymerase, OGG1 = 8-oxoguanine glycosylase, XRCC1 = X-ray repair cross-complementing protein 1, Pol β = DNA polymerase beta, PCNA = proliferating cell nuclear antigen, FEN1 = flap endonuclease 1, ATM = ataxia telangiectasia, mutated, MRN complex = Mre11 + RAD50 + NBS1/nibrin, RPA = replication protein A, BRCA2 = FANCD1 breast cancer susceptibility gene and DNA repair enzyme, Pol δ = DNA polymerase delta, Pol ε = DNA polymerase sigma, KU70/80 = lupus Ku autoantigen protein p70/p80, DNA PKcs = DNA-dependent protein kinase, catalytic subunit, XRCC4 = X-ray repair cross-complementing protein 4, XLF = XRCC4-like factor, and Pol μ = DNA polymerase mu. Created with BioRender.com; accessed January 21, 2021.
PARP was initially identified as a potential therapeutic target in SCLC through seminal work by Byers et al., who performed unbiased proteomic analysis of cell lines using reverse-phase protein arrays (RPPA) to identify proteins that were differentially expressed in SCLC compared with non-small-cell lung carcinoma (NSCLC) [21]. PARP1 transcript and protein levels were significantly elevated in SCLC cell lines compared to NSCLC. Increased PARP1 protein expression was also confirmed by immunohistochemical (IHC) analysis of tissue microarrays. Notably, several other components of the DDR pathway were increased in SCLC, including the checkpoint kinases CHK1 and CHK2, the ataxia telangiectasia related protein ATR, and the DNA-dependent protein kinase catalytic subunit DNA PKcs, which may be important to maintain cell viability in light of high replication stress (Figure 1). Treatment of a series of lung cancer cell lines with AZD2281 (olaparib) demonstrated that SCLC lines were significantly more sensitive to PARP inhibition than other histologic subtypes of lung cancer. Combining PARP inhibition with chemotherapy further decreased tumor cell viability.
These observations led to the initial studies of PARP inhibitors in SCLC as single agents (Table 1). In a phase I trial of talazoparib, 23 patients with SCLC were treated at the recommended phase II dose of 1.0 mg daily [30]. Two patients had a partial response, for an objective response rate (ORR) of 9% with a duration of response of 12.0 and 15.3 weeks. Both patients with an objective response had a platinum-free interval of 6 months or less. An additional four patients had stable disease, for a clinical benefit rate of 26% at 16 weeks. The UK STOMP trial examined the role of olaparib in the maintenance setting, but failed to show an improvement in progression-free survival (PFS) [31].
Table 1. Studies including PARP inhibitors in SCLC with outcomes data.
|
Study Population |
Drug(s) |
Response Rate |
PFS (months) |
OS (months) |
Unique Trial Data |
|
Patients with ≤1 prior treatment regimen30 |
Talazoparib |
9% |
11.1 weeks |
|
|
|
First-line ES-SCLC32 |
CE + veliparib vs. CE + placebo |
71.9% vs. 65.6% |
6.1 vs. 5.5 |
10.3 vs. 8.9 |
Elevated LDH and male gender correlated with benefit |
|
First-line ES-SCLC45 |
(A) CE+ veliparib -> veliparib (B) CE + veliparib -> placebo (C) CE + placebo -> placebo |
77% 59.3% 63.9% |
5.8 5.7 5.6 |
10.1 10.0 12.4 |
|
|
Relapsed ES-SCLC36 |
TMZ + veliparib vs. TMZ + placebo |
39% vs. 14% |
3.8 vs. 2.0 |
8.2 vs. 7.0 |
SLFN11 positive tumors prolonged PFS and OS |
|
Relapsed ES-SCLC39 |
TMZ + olaparib |
41.7% |
4.2 |
8.5 |
Co-clinical PDX trial |
|
Relapsed ES-SCLC65 |
Durvalumab + olaparib |
10.5% |
1.8 |
4.1 |
Inflamed phenotypeà response |
|
Relapsed ES-SCLC66 |
Durvalumab + olaparib |
5.3% |
|
|
Olaparib run in |
PFS = progression-free survival, OS = overall survival, ES-SCLC = extensive-stage small-cell lung cancer, CE = cisplatin/etoposide, LDH = lactate dehydrogenase, TMZ = temozolamide, SLFN11 = schlafen family member 11, and PDX = patient-derived xenograft.
This entry is adapted from the peer-reviewed paper 10.3390/cancers13040727