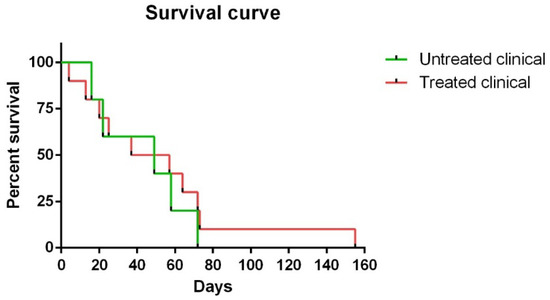
Neuroinflammation has been correlated with the progress of neurodegeneration in many neuropathologies. Although glial cells have traditionally been considered to be protective, the concept of them as neurotoxic cells has recently emerged. Thus, a major unsolved question is the exact role of astroglia and microglia in neurodegenerative disorders. On the other hand, it is well known that glucocorticoids are the first choice to regulate inflammation and, consequently, neuroglial inflammatory activity. The objective of this study was to determine how chronic dexamethasone treatment influences the host immune response and to characterize the beneficial or detrimental role of glial cells. To date, this has not been examined using a natural neurodegenerative model of scrapie. With this aim, immunohistochemical expression of glial markers, prion protein accumulation, histopathological lesions and clinical evolution were compared with those in a control group. Although impact of dexamethasone administration on neuropathological lesions was not demonstrated and treatment did not seem to be clinically relevant to disease progress when clinical signs had already begun, the evident extension of survival in one case was hopeful. The findings presented in this study support a potential failure of astrocytes and a stimulation of phagocytosis of PrPsc deposits by microglia. Thus, it is evidenced here how the complex interaction between glial populations failed to compensate for brain damage in natural conditions, emphasizing the need for using natural models. Additionally, the data showed that modulation of neuroinflammation by anti-inflammatory drugs might become a research focus as a potential therapeutic target for prion diseases, similar to that considered previously for other neurodegenerative disorders classified as prion-like diseases.
- dexamethasone
- neuroinflammation
- astrocytes
- microglia
- prion diseases
- neurodegeneration
Results
Clinical Signs
| Group | Sheep No | Main Clinical Signs |
|---|---|---|
| Clinical non-treated | 11 | Tremors, pruritus, ataxia, lost look |
| 12 | Pruritus with skin lesions, alopecia, hyperexcitation | |
| 13 | Pruritus, alopecia, prostration | |
| 14 | Tremors, alopecia, prostration | |
| 15 | Pruritus | |
| Clinical DEX-treated | 16 | Pruritus, ataxia, tremors |
| 17 | Pruritus, alopecia | |
| 18 | Tremors, ataxia, hyperexcitation | |
| 19 | Pruritus, alopecia, hyperexcitation | |
| 20 | Tremors, intense widespread alopecia, prostration | |
| 21 | Hyperexcitation, prostration | |
| 22 | Tremors, constant pruritus, bruxism | |
| 23 | Scarce pruritus | |
| 24 | Intense pruritus with skin lesions | |
| 25 | Pruritus, alopecia, prostration |
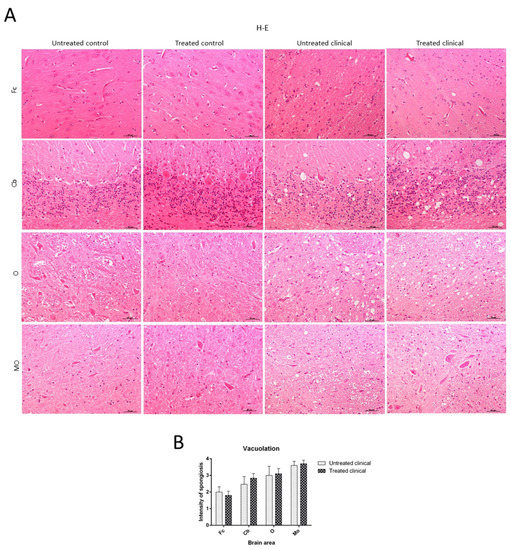
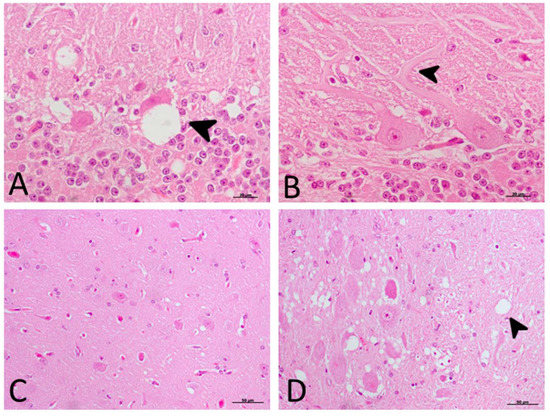
Histopathological Findings (H-E)
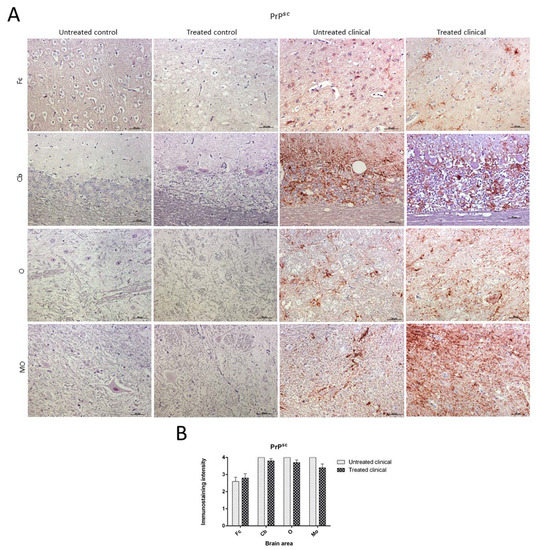
Immunohistochemical (IHC) Findings
PrPsc Accumulation
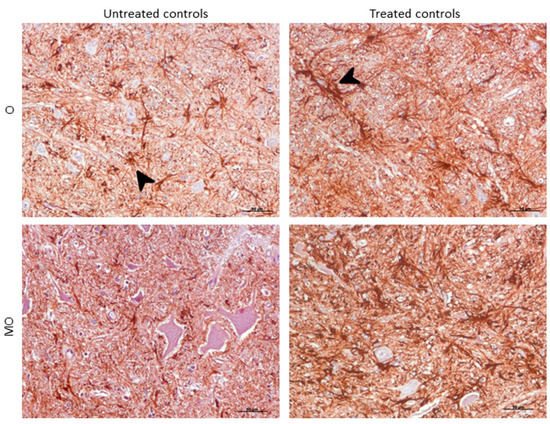
GFAP
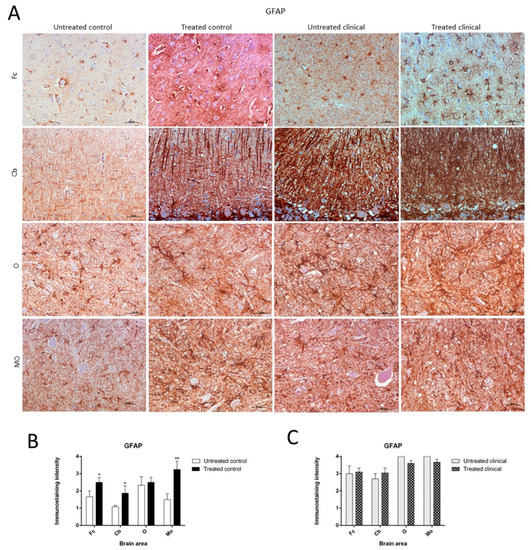
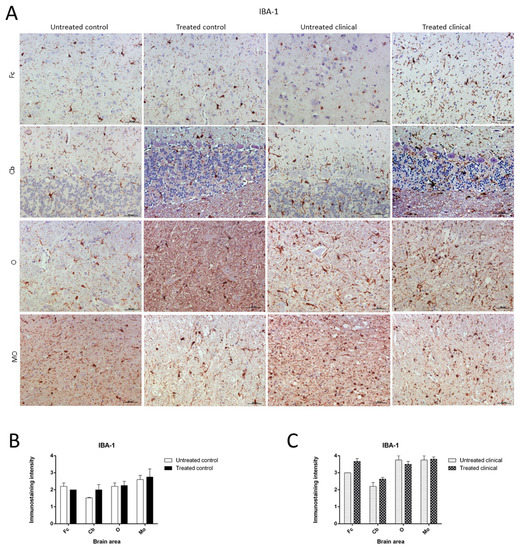
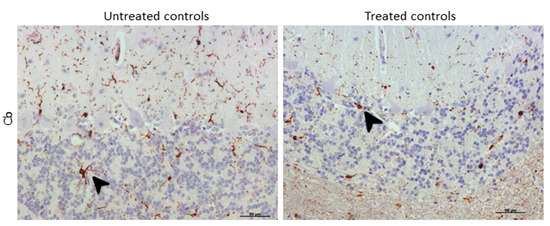
IBA-1
RT-qPCR
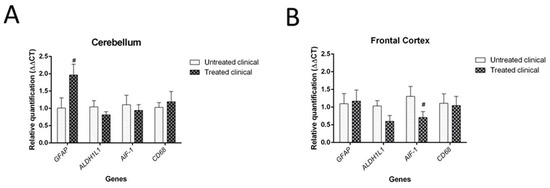
Discussion
- Kimberlin, R.H.; Walker, C.A. The antiviral compound HPA-23 can prevent scrapie when administered at the time of infection. Arch. Virol. 1983, 78, 9–18. [Google Scholar] [CrossRef]
- White, A.R.; Enever, P.; Tayebi, M.; Mushens, R.; Linehan, J.; Brandner, S.; Anstee, D.; Collinge, J.; Hawke, S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003, 422, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Grilli, M.; Memo, M. Nuclear factor-kappaB/Rel proteins, a point of convergence of signalling pathways relevant in neuronal function and dysfunction. Biochem. Pharmacol. 1999, 57, 1–7. [Google Scholar] [CrossRef]
- Efremova, L.; Chovancova, P.; Adam, M.; Gutbier, S.; Schildknecht, S.; Leist, M. Switching from astrocytic neuroprotection to neurodegeneration by cytokine stimulation. Arch. Toxicol. 2017, 91, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Kurkowska-Jastrzebska, I.; Litwin, T.; Joniec, I.; Ciesielska, A.; Przybylkowski, A.; Czlonkowski, A.; Czlonkowska, A. Dexamethasone protects against dopaminergic neurons damage in a mouse model of Parkinson’s disease. Int. Immunopharmacol. 2004, 4, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Nerius, M.; Haenisch, B.; Gomm, M.; Doblhammer, G.; Schneider, A. Glucocorticoid Therapy is Associated with a Lower Risk of Dementia. J. Alzheimers Dis. 2020, 73, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Green, K.N.; Billings, L.M.; Roozendaal, B.; McGaugh, J.L.; LaFerla, F.M. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 2006, 26, 9047–9056. [Google Scholar] [CrossRef] [PubMed]
- Outram, G.W.; Dickinson, A.G.; Fraser, H. Reduced susceptibility to scrapie in mice after steroid administration. Nature 1974, 249, 855–856. [Google Scholar] [CrossRef]
- Riemer, C.; Burwinkel, M.; Schwarz, A.; Gultner, S.; Mok, S.W.F.; Heise, I.; Holtkamp, N.; Baier, M. Evaluation of drugs for treatment of prion infections of the central nervous system. J. Gen. Virol. 2008, 89, 594–597. [Google Scholar] [CrossRef]
- Imbimbo, B.P. An update on the efficacy of non-steroidal anti-inflammatory drugs in Alzheimer’s disease. Expert Opin. Investig. Drugs 2009, 18, 1147–1168. [Google Scholar] [CrossRef]
- Tischner, D.; Reichardt, H.M. Glucocorticoids in the control of neuroinflammation. Mol. Cell Endocrinol. 2007, 275, 62–70. [Google Scholar] [CrossRef]
- Min, L.; Hodi, F.S.; Kaiser, U.B. Corticosteroids and immune checkpoint blockade. Aging (Albany NY) 2015, 7, 521–522. [Google Scholar] [CrossRef] [PubMed]
- Munck, A.; Naray-Fejes-Toth, A. The ups and downs of glucocorticoid physiology. Permissive and suppressive effects revisited. Mol. Cell Endocrinol. 1992, 90, C1–C4. [Google Scholar] [CrossRef]
- Filaretova, L.; Podvigina, T.; Bagaeva, T.; Morozova, O. Dual action of glucocorticoid hormones on the gastric mucosa, how the gastroprotective action can be transformed to the ulcerogenic one. Inflammopharmacology 2009, 17, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Sorrells, S.F.; Caso, J.R.; Munhoz, C.D.; Sapolsky, R.M. The stressed CNS, when glucocorticoids aggravate inflammation. Neuron 2009, 64, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Spies, C.M.; Strehl, C.; van der Goes, M.C.; Bijlsma, J.W.; Buttgereit, F. Glucocorticoids. Best Pract Res. Clin. Rheumatol. 2011, 25, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, J.R. Glucocorticoid resistance in patients with rheumatoid arthritis. Scand. J. Rheumatol. 2007, 36, 165–166. [Google Scholar] [CrossRef]
- De Kloet, E.R.; Vreugdenhil, E.; Oitzl, M.S.; Joels, M. Brain corticosteroid receptor balance in health and disease. Endocr. Rev. 1998, 19, 269–301. [Google Scholar] [CrossRef]
- Meijer, O.C.; de Lange, E.C.; Breimer, D.D.; de Boer, A.G.; Workel, J.O.; de Kloet, E.R. Penetration of dexamethasone into brain glucocorticoid targets is enhanced in mdr1A P-glycoprotein knockout mice. Endocrinology 1998, 139, 1789–1793. [Google Scholar] [CrossRef]
- Hueston, C.M.; Deak, T. The inflamed axis, the interaction between stress, hormones, and the expression of inflammatory-related genes within key structures comprising the hypothalamic-pituitary-adrenal axis. Physiol. Behav. 2014, 124, 77–91. [Google Scholar] [CrossRef]
- Giles, A.J.; Hutchinson, M.N.D.; Sonnemann, H.M.; Jung, J.; Fecci, P.E.; Ratnam, N.M.; Zhang, W.; Song, H.; Bailey, R.; Davis, D.; et al. Dexamethasone-induced immunosuppression, mechanisms and implications for immunotherapy. J. Immunother. Cancer 2018, 6, 51. [Google Scholar] [CrossRef]
- Weindl, G.; Schaller, M.; Schafer-Korting, M.; Korting, H.C. Hyaluronic acid in the treatment and prevention of skin diseases, molecular biological, pharmaceutical and clinical aspects. Skin Pharmacol. Physiol. 2004, 17, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, M.W. Anti-inflammatory glucocorticoid drugs, reflections after 60 years. Inflammopharmacology 2011, 19, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sarna, J.R.; Hawkes, R. Patterned Purkinje cell death in the cerebellum. Prog. Neurobiol. 2003, 70, 473–507. [Google Scholar] [CrossRef]
- Sarasa, R.; Junquera, C.; Toledano, A.; Badiola, J.J.; Monzón, M. Ultrastructural changes in the progress of natural Scrapie regardless fixation protocol. Histochem. Cell Biol. 2015, 144, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Lezmi, S.; Seuberlich, T.; Oevermann, A.; Baron, T.; Bencsik, A. Comparison of brain PrPd distribution in ovine BSE and scrapie. Vet. Pathol. 2011, 48, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Alibhai, J.; Blanco, R.A.; Barria, M.A.; Piccardo, P.; Caughey, B.; Perry, V.H.; Freeman, T.C.; Manson, J.C. Distribution of Misfolded Prion Protein Seeding Activity Alone Does Not Predict Regions of Neurodegeneration. PLoS Biol. 2016, 14, e1002579. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Marutle, A.; Rodriguez-Arellano, J.J.; Nordberg, A. Glial Asthenia and Functional Paralysis, A New Perspective on Neurodegeneration and Alzheimer’s Disease. Neuroscientist 2015, 21, 552–568. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Rodrigues, J.J.; Pivoriunas, A.; Zorec, R.; Semyanov, A. Astroglial atrophy in Alzheimer’s disease. Pflugers Arch. 2019, 471, 1247–1261. [Google Scholar] [CrossRef]
- Fraser, H.; Brown, K.L.; Stewart, K.; McConnell, I.; McBride, P.; Williams, A. Replication of scrapie in spleens of SCID mice follows reconstitution with wild-type mouse bone marrow. J. Gen. Virol. 1996, 77, 1935–1940. [Google Scholar] [CrossRef]
- Berciano, J.; Berciano, M.T.; Polo, J.M.; Figols, J.; Ciudad, J.; Lafarga, M. Creutzfeldt-Jakob disease with severe involvement of cerebral white matter and cerebellum. Virchows Arch. Pathol. Anat. Histopathol. 1990, 417, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A.; Ironside, J.W.; Lantos, P.L.; Cairns, N.J. A quantitative study of the pathological changes in the cerebellum in 15 cases of variant Creutzfeldt-Jakob disease (vCJD). Neuropathol. Appl. Neurobiol. 2009, 35, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Cali, I.; Miller, C.J.; Parisi, J.E.; Geschwind, M.D.; Gambetti, P.; Schonberger, L.B. Distinct pathological phenotypes of Creutzfeldt-Jakob disease in recipients of prion-contaminated growth hormone. Acta. Neuropathol. Commun. 2015, 3, 37. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.I.; Rivas, L.; Lacruz, C.; Toledano, A. Astroglial cell subtypes in the cerebella of normal adults, elderly adults, and patients with Alzheimer’s disease, a histological and immunohistochemical comparison. Glia 2015, 63, 287–312. [Google Scholar] [CrossRef] [PubMed]
- Zeug, A.; Muller, F.E.; Anders, S.; Herde, M.K.; Minge, D.; Ponimaskin, E.; Henneberger, C. Control of astrocyte morphology by Rho GTPases. Brain Res. Bull. 2018, 136, 44–53. [Google Scholar] [CrossRef]
- Perry, V.H.; Cunningham, C.; Boche, D. Atypical inflammation in the central nervous system in prion disease. Curr. Opin. Neurol. 2002, 15, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; O’Connor, V. The role of microglia in synaptic stripping and synaptic degeneration, a revised perspective. ASN Neuro 2010, 2, e00047. [Google Scholar] [CrossRef]
- Meneses, G.; Gevorkian, G.; Florentino, A.; Bautista, M.A.; Espinosa, A.; Acero, G.; Diaz, G.; Fleury, A.; Perez Osorio, I.N.; Del Rey, A.; et al. Intranasal delivery of dexamethasone efficiently controls LPS-induced murine neuroinflammation. Clin. Exp. Immunol. 2017, 190, 304–314. [Google Scholar] [CrossRef]
- Hui, B.; Yao, X.; Zhang, L.; Zhou, Q. Dexamethasone sodium phosphate attenuates lipopolysaccharide-induced neuroinflammation in microglia BV2 cells. Naunyn Schmiedebergs Arch. Pharmacol. 2020. [CrossRef]
- Duque Ede, A.; Munhoz, C.D. The Pro-inflammatory Effects of Glucocorticoids in the Brain. Front. Endocrinol (Lausanne) 2016, 7, 78. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology, unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Boche, D.; Perry, V.H.; Nicoll, J.A. Review, activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Xue, Q.S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142. [Google Scholar] [CrossRef]
- Kim, S.U.; de Vellis, J. Microglia in health and disease. J. Neurosci. Res. 2005, 81, 302–313. [Google Scholar] [CrossRef]
- Muhleisen, H.; Gehrmann, J.; Meyermann, R. Reactive microglia in Creutzfeldt-Jakob disease. Neuropathol. Appl. Neurobiol. 1995, 21, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Lopez-Gonzalez, I.; Thune, K.; Carmona, M.; Zafar, S.; Andreoletti, O.; Zerr, I.; Ferrer, I. Subtype and regional-specific neuroinflammation in sporadic creutzfeldt-jakob disease. Front. Aging. Neurosci. 2014, 6, 198. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Trojanowski, P.J.; Villanueva, E.; Navarro, E.; Godbout, J.P. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 2016, 64, 300–316. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation, the devil is in the details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef]
- Obst, J.; Simon, E.; Mancuso, R.; Gomez-Nicola, D. The Role of Microglia in Prion Diseases, A Paradigm of Functional Diversity. Front. Aging Neurosci. 2017, 9, 207. [Google Scholar] [CrossRef]
- Buffo, A.; Rolando, C.; Ceruti, S. Astrocytes in the damaged brain, molecular and cellular insights into their reactive response and healing potential. Biochem. Pharmacol. 2010, 79, 77–89. [Google Scholar] [CrossRef]
- Garwood, C.J.; Pooler, A.M.; Atherton, J.; Hanger, D.P.; Noble, W. Astrocytes are important mediators of Abeta-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis. 2011, 2, e167. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes, Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Kirkley, K.S.; Popichak, K.A.; Afzali, M.F.; Legare, M.E.; Tjalkens, R.B. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J. Neuroinflammation 2017, 14, 99. [Google Scholar] [CrossRef]
- Espinosa, J.C.; Nonno, R.; Di Bari, M.; Aguilar-Calvo, P.; Pirisinu, L.; Fernandez-Borges, N.; Vanni, I.; Vaccari, G.; Marin-Moreno, A.; Frassanito, P.; et al. PrPC Governs Susceptibility to Prion Strains in Bank Vole, While Other Host Factors Modulate Strain Features. J. Virol. 2016, 90, 10660–10669. [Google Scholar] [CrossRef]
- Mathiason, C.K. Scrapie, CWD, and Transmissible Mink Encephalopathy. Prog. Mol. Biol. Transl. Sci. 2017, 150, 267–292. [Google Scholar]
- Kriz, J.; Nguyen, M.D.; Julien, J.P. Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2002, 10, 268–278. [Google Scholar] [CrossRef]
- Choi, J.K.; Jenkins, B.G.; Carreras, I.; Kaymakcalan, S.; Cormier, K.; Kowall, N.W.; Dedeoglu, A. Anti-inflammatory treatment in AD mice protects against neuronal pathology. Exp. Neurol. 2010, 223, 377–384. [Google Scholar] [CrossRef]
- Burwinkel, M.; Riemer, C.; Schwarz, A.; Schultz, J.; Neidhold, S.; Bamme, T.; Baier, M. Role of cytokines and chemokines in prion infections of the central nervous system. Int. J. Dev. Neurosci. 2004, 22, 497–505. [Google Scholar] [CrossRef]
This entry is adapted from the peer-reviewed paper 10.3390/ijms21093231