Endometrial cancer (EC) is one of the most common gynecological malignancies. Gonadotropin releasing hormone (GnRH) is a decapeptide first described to be secreted by the hypothalamus to regulate pituitary gonadotropin secretion. In this systematic review, we analyze and summarize the data indicating that most EC express GnRH and its receptor (GnRH-R) as part of an autocrine system regulating proliferation, the cell cycle, and apoptosis. We analyze the available data on the expression and function of GnRH-II, its putative receptor, and its signal transduction. GnRH-I and GnRH-II agonists, and antagonists as well as cytotoxic GnRH-I analogs, have been shown to inhibit proliferation and to induce apoptosis in human EC cell lines in pre-clinical models. Treatment with conventional doses of GnRH-agonists that suppress pituitary gonadotropin secretion and ovarian estrogen production has become part of fertility preserving therapy of early EC or its pre-cancer (atypical endometrial hyperplasia). Conventional doses of GnRH-agonists had marginal activity in advanced or recurrent EC. Higher doses or more potent analogs including GnRH-II antagonists have not yet been used clinically. The cytotoxic GnRH-analog Zoptarelin Doxorubicin has shown encouraging activity in a phase II trial in patients with advanced or recurrent EC, which expressed GnRH-R. In a phase III trial in patients with EC of unknown GnRH-R expression, the cytotoxic GnRH doxorubicin conjugate was not superior to free doxorubicin. Further well-designed clinical trials exploiting the GnRH-system in EC might be useful.
- endometrial cancer
- gonadotropin releasing hormone
- luteinizing hormone releasing hormone
- GnRH
- LHRH
1. Introduction
Endometrial cancer (EC), derived from the epithelial lining of the uterine cavity, is one of the most common female cancers. Worldwide, 382,069 new cases were diagnosed in 2018 and 89,929 women died of this disease [1]. In Western Europe and North America, EC is the fourth most common malignancy in women and the most common malignant tumor of the female genital organs [2].
The prognosis of EC is rather favorable, as about 75% of cases are diagnosed in an early stage and can be cured by surgery and in more advanced cases by additional radiotherapy and/or chemotherapy [2] resulting in a general five-year cancer-specific survival of around 80% for all stages and histological types [1,2]. The majority of EC (85%) known as the so-called type 1 cancers develop due to prolonged exposure to endogenous or exogenous estrogens in the absence of sufficient progestogen activity [3,4,5]. These type 1 EC are hormone-dependent and can be treated in early stages without surgery by endocrine manipulation, including estrogen withdrawal and/or high dose progestogens in premenopausal women who have not yet completed their families. Later in their development, these hormone-dependent EC dedifferentiate, lose expression of estrogen receptors and/or progesterone receptors, and are no longer amenable to estrogen withdrawal, anti-estrogens, or progestogens [2]. Type 2 EC do not express estrogen receptors or progesterone receptors, are not dependent on these steroids, and have a poor prognosis. They are responsible for the majority of EC-related deaths [2].
Gonadotropin releasing hormone (GnRH, also called luteinizing hormone releasing hormone, LHRH) is a hypothalamic decapeptide, regulating secretion of gonadotropins by the pituitary. By the end of the 1980s, super-active analogs of GnRH and respective depot preparations became widely available and were used for medical hypophysectomy (suppression of secretion of luteinizing hormone, LH, and follicle-stimulating hormone, FSH) leading to suppression of gonadal function in both sexes. This strategy of reversible medical castration was successfully introduced into the treatment of a variety of sex hormone-dependent diseases including prostate cancer and premenopausal breast cancer as well as endometriosis and uterine fibroids [6]. Estrogen withdrawal due to reversible medical castration through GnRH-agonists was also established as a conservative treatment of early EC and its pre-cancers in young women who wished to preserve their fertility [6].
In a variety of malignant tumors including breast, prostate, ovarian, and endometrial cancers, the expressions of GnRH and its receptors (GnRH-R) were discovered. These GnRH-R mediated direct anti-proliferative effects of agonistic and antagonistic analogs of GnRH in vitro and in nude mouse models xeno-transplanted with human cancers. Since GnRH-R are also expressed in type 2 EC, it was speculated that the treatment with GnRH-analogs might be an efficacious endocrine therapy with low toxicity for patients with EC that does not express estrogen receptors or progesterone receptors [6]. Respective clinical trials showed some activity of this approach. Finally, GnRH-analogs coupled with cytotoxic molecules were developed for targeted therapies through the GnRH-R on the surface of cancer cells. This approach has provided encouraging results in patients with EC (see below).
2. Discussion
2.1. GnRH-Receptors in EC
The data reviewed above clearly suggest that most human EC cell lines and primary EC express high affinity/low-capacity receptors for GnRH. It is true that some groups were not able to find these GnRH-R [10,12,14]. However, most researchers worldwide detected them either by binding assay, RT-PCR, restriction enzyme analysisand Southern blot analysis or immunohistochemistry [7,9,11,13,15,16,17,19,20,21,22,23,24,25,26,27,28]. The sequence of the cDNA of the GnRH-R in HEC-1A and Ishikawa human EC cell lines is identical to that of the GnRH-R in a human pituitary gland [18]. Approximately 50–95% of primary human EC express GnRH-R [7,11,17,19,20,23,24,25,28].
2.2. Expression of GnRH in EC
The production of GnRH by EC-cell lines and in 75–100% of primary tumor samples was shown by immunoassay, bioassay, high performance liquid chromatography, immunohistochemistry, RT-PCR, restriction enzyme analysis, and Southern blot analysis [12,24,29,30], so that this finding is also well accepted.
2.3. Effects of GnRH, GnRH-Agonists, and GnRH Antagonists on the Proliferation of EC Cells
In established human EC cell lines as well as in primary cultures of EC, most researchers found a growth inhibition induced by treatment with GnRH-agonists [9,13,14,15,17,20,34,35]. Comparable anti-proliferative effects were seen, when GnRH-antagonists were used [9,14,17,31]. Under serum free conditions, IGF-1, EGF, and estrogen induced proliferation of EC cell lines was reduced by GnRH analogs [31,32,33]. Significant anti-proliferative effects of GnRH analogs were seen at 10 pM to 1 nM concentrations [9], but relevant growth inhibition was observed at higher concentrations (1 µM, 10 µM) [9,13,14,15,17,20,32,33,34,35].
A common finding made by all groups was that GnRH-antagonists had the same anti-proliferative effects on EC cells as GnRH-agonists, suggesting that the dichotomy between GnRH-agonists and antagonists, known from the pituitary, is not valid in EC cells [6]. To elucidate the effects of GnRH produced and secreted by the tumor cells, we treated cell cultures of human ovarian cancer cell lines EFO-21 and EFO-27 that had been shown to express high affinity GnRH-R and to secrete GnRH with neutralizing concentrations of antiserum to GnRH. This resulted in a significant stimulation of proliferation. Native GnRH in low concentrations had no or little anti-proliferative effect [36].
Thus, it seems reasonable to conclude that the majority of human EC express GnRH and GnRH-R as an autocrine system reducing their proliferation. Both GnRH agonists and antagonists have dose-dependent anti-proliferative effects.
2.4. Signal Transduction and Intracellular Actions of GnRH in EC
In the pituitary gonadotrope GnRH-R that have bound the GnRH couple to G-protein αq and induce activation of phospholipase (PLC), protein kinase (PKC), and adenylyl cyclase (AC) [12,14,32]. These enzymes and pathways are present in human EC cell lines but are not involved in the mediation of anti-proliferative effects of GnRH agonists [32]. Upon binding of a GnRH agonist or antagonist, the GnRH-R rather couples to G-protein αi and activates a phosphotyrosine phosphatase that reduces EGF induced tyrosine phosphorylation of the EGF-R [18,32,38,44,45,46]. This results in a suppression of the Ras/MAPK/ERK pathway and an inhibition of c-fos expression [18,32,38] (Figure 1). This mechanism has been shown in human breast and ovarian cancer cell lines [86]. In addition, through this mechanism, GnRH-agonists can inhibit E2 induced cell proliferation of ERα-positive human endometrial, ovarian, and breast cancer cell lines (Figure 1) [40]. In breast cancers, signalling of membrane-bound G-Protein-coupled estrogen receptor 1 (GPER) through transactivation of EGF-R could also be inhibited by GnRH-agonist treatment (Figure 1) [86]. Thus, E2-induced proliferation of ERα-negative but GPER-positive breast cancer cells could be prevented by treatment with GnRH agonists.
GnRH agonists stimulated via G-protein αi the JNK/AP-1 pathway, leading to inhibition of the cell cycle (Figure 1) [39]. Native GnRH, GnRH-agonists, and GnRH-antagonists did not induce apoptosis but rather protected endometrial cancer cells from apoptosis induction by UV-light or the cytotoxic agent doxorubicin through activation of NF-κB (Figure 1) [42]. Thus, we conclude from our data and those of others, that the expression of GnRH and GnRH-R in human EC is part of an autocrine system, counteracting the proliferative effects of growth factors and estrogens, and increasing AP-1 expression, leading to cell cycle arrest. In addition, this GnRH system reduces apoptosis (Figure 1). Other authors, however, found induction of apoptosis caused by GnRH-agonists and/or antagonists, the activation of protein kinase C, and other mechanisms including FasL and FAS, telomerase transferase, telomerase activity, and annexin V [13,15,16,20,34,35,46,47].
The reasons for these discrepancies are unknown. It is not unreasonable to speculate that the GnRH-R in tumor cells can couple to multiple signal-transduction pathways depending on the cellular milieu in different cell lines and even different passages of the same cell line [86].
2.5. GnRH-II and GnRH-II-Receptors in EC
The data from our group suggest that a truncated, but functional 5-transmembrane human GnRH-II-R, is expressed in human endometrial and ovarian cancers. After binding of the GnRH-antagonist, native GnRH-II, or GnRH-II-agonist, it couples to G protein αi and activates the signal transduction mechanisms described for the GnRH-I-R in human cancers including inhibition of autophosphorylation of the EGF-receptor and phosphorylation of JNK [54,55,57,58] (Figure 2). Our findings suggest that the antiproliferative effects of GnRH-I antagonists are mediated through GnRH-II-R. The anti-proliferative effects of native GnRH-II mediated through GnRH-II-R were much more pronounced than those of GnRH-I agonist mediated through GnRH-I-R [54,55,56,57,58].
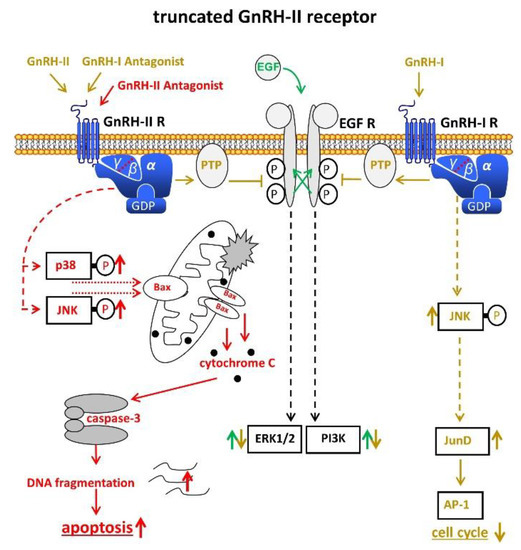
Figure 2. Signal transduction of the putative truncated GnRH-II receptor in endometrial cancer (EC). Binding of GnRH-II, GnRH-II agonists, and GnRH-I antagonists cause activation of PTP, leading to dephopsphorylation of activated EGF-R with the consequences described in Figure 1. Binding of GnRH-II antagonists induce apoptosis through activation of p38, JNK, and the intrinsic apoptotic pathway. AP-1, activvator protein-1. Bax, B-cell lymphoma 2 (Bcl-2)-associated X protein. EGF, epidermal growth factor. EGF-R, EGF receptor. ERK1/2, p44/42 mitogen-activated protein (MAP) kinase. GDP, guanosindiphosphat. GnRH, gonadotropin releasing hormone. JNK, c-Jun N-terminale kinase. JunD, transcription factor JunD. P38, mitogen-activated protein kinase P38. PI3K, phosphoinositide 3-kinase. PTP, protein tyrosine phosphatase.
GnRH-I, GnRH-I-agonists, GnRH-I-antagonists, and GnRH-II-agonists did not increase apoptosis in EC, ovarian, or breast cancer cell lines, but rather protected cells from programmed cell death [58,59]. Antagonists of GnRH-II, however, potently induced apoptosis in human endometrial, ovarian, and breast cancer cells, mediated through activation of stress-induced mitogen activated protein kinases p38 and c-JunNH2-terminal kinase, leading to activation of proapoptotic protein Bax [58,59] (Figure 2). GnRH-II antagonists potently inhibited the growth of human endometrial and ovarian cancers in nude mice [58].
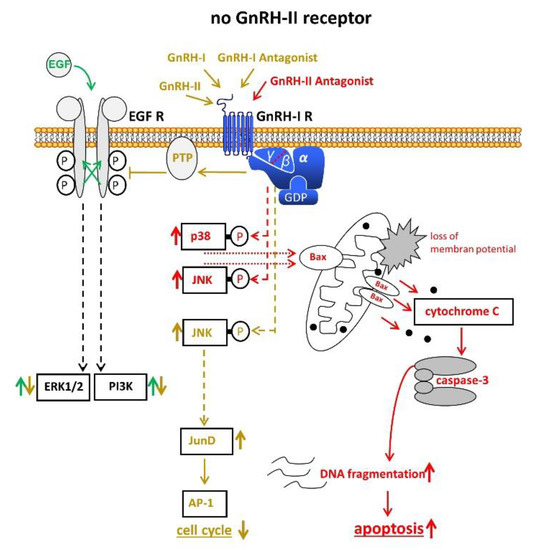
Figure 3. Signal transduction of GnRH-I and GnRH-II and their agonistic as well as antagonistic analogs through the GnRH-I-R if no functional GnRH-II-R is expressed. For details, cf. Figure 1 and Figure 2. AP-1, activvator protein-1. Bax, B-cell lymphoma 2 (Bcl-2)-associated X protein. EGF, epidermal growth factor. EGF-R, EGF receptor. ERK1/2, p44/42 mitogen-activated protein (MAP) kinase. GDP, guanosindiphosphat. GnRH, gonadotropin releasing hormone. JNK, c-Jun N-terminale kinase. JunD, transcription factor JunD. P38, mitogen-activated protein kinase P38. PI3K, phosphoinositide 3-kinase. PTP, protein tyrosine phosphatase.
2.6. GnRH-R in EC as a Target for Cytotoxic Therapy
The in vitro and in vivo experimental data available suggest that GnRH-R can be utilized as a target to specifically introduce toxic molecules into EC cells [21,61,62,63,64,65]. Of particular interest is the finding that cytotoxic GnRH-molecules can bypass the multi-drug resistance 1 system that eventually makes tumor cells refractory to chemotherapy [64].
2.7. Clinical Application of GnRH Analogs in EC and Atypical Endometrial Hyperplasia (AEH)
Estrogen withdrawal by the GnRH-agonist induced reversible medical castration, which has been used in combination with systemic or local progestogen application as a fertility sparing treatment for AEH and early EC [69,70,71,72,73,76]. Alternatively, GnRH agonists were combined with aromatase inhibitors for more intensive suppression of estrogens [73,74,76] or given alone after complete endometrial resection [75]. In these small series, complete response rates of about 70% (54–100%) were observed for early EC. Complete response rates were higher in AEH (80–100%). Pregnancy rates were about 50%. A relevant number of patients had progression during therapy or relapsed after complete remission [69,70,71,72,73,74,75,76]. Estrogen withdrawal through reversible medical castration by GnRH analogs alone or in combination with a LNG-IUD or an aromatase inhibitor seems to be preferable in obese patients [76] (Table 1).
A series of small phase II trials has assessed the efficacy of conventional doses of GnRH-agonists in the treatment of recurrent or advanced EC no longer amenable to surgery or radiotherapy, including patients with G3 tumors. Objective responses were observed in 0% to 35%, which tended to be long lasting. Toxicity was low [77,78,79,80,81,82]. It has to be considered that the serum concentrations of GnRH-agonists achieved with standard depot-preparations are in the nanomolar range and sufficient to down-regulate pituitary GnRH-R [6]. For optimal direct effects of GnRH-I-agonists or antagonists on EC cells, much higher concentrations (µM) are needed [9,13,14,15,17,20,31,32,34,35]. Clinical trials with higher doses of GnRH-I-analogs have not been performed. GnRH-II analogs, especially GnRH-II antagonists that are much more potent in vitro and in vivo [58,59], should be tested in EC patients.
Phase I and II trials with the cytotoxic GnRH analog Zoptarelin Doxorubicin resulted in an objective response rate of 23% and a clinical benefit rate of 67% in patients with advanced or recurrent EC that expressed GnRH-R [25]. Toxicity was low. In a phase III trial with patients with recurrent or metastatic EC that had failed to prior platinum chemotherapy, an equipotency of Zoptarelin Doxorubicin and free Doxorubicin was found. However, the GnRH-R-status of these tumors was unknown. Considering that Zoptarelin Doxorubicin had been shown to be ineffective in EC not expressing GnRH-R [61,62,63] and that 5–50% of EC have no GnRH-R [20,23,24,25], it is reasonable to speculate that the Zoptarelin Doxorubicin is more efficacious in patients with EC that express GnRH-R.
3. Conclusions and Perspectives
Treatment with conventional doses of GnRH-agonists that suppress pituitary gonadotropin secretion and ovarian estrogen production has become part of fertility preserving therapy either alone or in combination with LNG-IUD (52 mg) or aromatase inhibitors in young patients with AEH or early EC (EEC).
There is convincing evidence that EC express GnRH-I-R and GnRH-I as a negative autocrine system, limiting cell proliferation and likely apoptosis. It is unclear whether a functional GnRH-II-R exist in humans. The GnRH-I-R and, if existent, the putative GnRH-II-R can be targeted by analogs of GnRH-I and GnRH-II to inhibit proliferation and to induce apoptosis. Clinical trials to exploit these direct anti-tumor effects have, so far, been performed with conventional doses of GnRH agonists, resulting in marginal efficacy, but low toxicity. Trials using higher doses of GnRH-I analogs or the more potent GnRH-II analogs still have to be performed. The cytotoxic GnRH-analog Zoptarelin Doxorubicin has been shown to have meaningful activity in animal models and a phase II trial. A well-designed phase III trial with patients with advanced or recurrent EC expressing GnRH-R is warranted.
This entry is adapted from the peer-reviewed paper 10.3390/cells10020292