Follicular Lymphoma (FL), the most common indolent non-Hodgkin’s B cell lymphoma, is a paradigm of the immune microenvironment’s contribution to disease onset, progression, and heterogeneity.
- follicular lymphoma
- microenvironment
- immunotherapy
1. FL Microenvironment: Friend or Foe?
Follicular lymphoma (FL), the most common indolent non-Hodgkin’s lymphoma (NHL), is a biologically heterogeneous disease with clinical variations in patient outcome [1].
The initial oncogenic hit happens in the Pre B/Pro B stage of B cells in the bone marrow (BM) where they acquire the t (14;18) translocation due to an error in V(D)J recombination. Subsequently, these cells home to B cell follicles inside lymph nodes (LNs) where they encounter the antigen (Ag) and undergo, in the germinal center (GC), somatic hypermutation (SHM) and class switch recombination (CSR) (IgM to IgG) of the immunoglobulin that constitutes the B cell receptor (BCR). FL-like cells interact with follicular dendritic cells (FDCs) and are selected to undergo apoptosis or be rescued by follicular helper T cells (TFH), based on the Ag affinity of their BCRs. Overexpression of BCL2, along with additional anti-apoptotic proteins, allow apoptosis escape independently of BCR affinity. These FL-like B cells then exit the GC and enter circulation where they might be prone to traffic between secondary lymphoid organs and the BM, and they acquire additional genetic changes necessary for transformation to FL, such as CREBBP, KMT2D, EZH2, TNFRS14, among others [2,3].
There is now growing evidence that crosstalk between lymphoma cells and stromal and immune cells in lymphoid compartments is fundamental for disease onset and progression. This crosstalk is dynamic and shapes the tumor microenvironment enhancing the pro-tumoral features of the niche [4,5]. FL represents a paradigm of dependence on the microenvironment. Seminal microarray studies in LN biopsies from the Leukemia and Lymphoma Profiling Project (LLMPP) series established for the first time that FL prognosis was not given by the tumor cell per se but by the composition of non-malignant cells [6–8]. Subsequently, many studies have tried to identify phenotypic markers to stratify patients, although this picture has been more complicated than anticipated, and influenced by treatment.
The main players that support tumors, through a complex set of cytokines, receptors, immune modulators, and pro-angiogenic factors, are follicular dendritic cells (FDCs), fibroblastic reticular cells (FRCs), mesenchymal stromal cells (MSCs), and tumor-associated macrophages (TMAs), together with a rich T cell infiltrate composed of CD4 T follicular helpers (TFH) cells, CD4 T follicular regulatory (TFR) cells, CD4 T regulatory cells (TREG), and CD8 cytotoxic T cells (CTL) [9,10] (Figure 1).
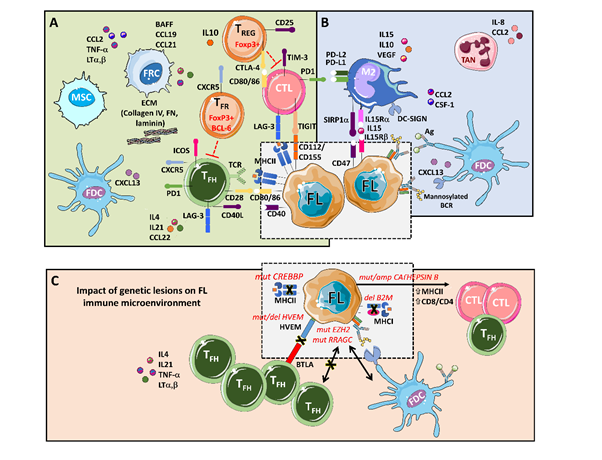
Figure 1. An integrative view of follicular lymphoma (FL) microenvironment and its crosstalk with genetic drivers. (A) FL is highly infiltrated with several T cell subpopulations, where TFH are fundamental players through MHC II and CD40L, while immunosuppressive TREG hamper cytotoxic T cells (CTLs) activation. Fibroblastic reticular cells (FRCs) also participate in immunosuppression by secreting extracellular matrix (ECM) proteins that regulate T cell trafficking and cooperate with TFH. (B) FL cells favor the recruitment of monocytes through CCL2 and CSF-1 that differentiate and polarize mostly into M2-like macrophages expressing PD-L1 and PD-L2 and dampening CTLs cytotoxic activity. Both macrophages and FDCs activate B cell receptors (BCRs) through lectins binding to mannosylated BCR. Likewise, FDCs also activate BCRs through the presentation of immunocomplexes to FL cells. Neutrophils are recruited through IL8 secretion in the FL niche and support lymphoma growth. (C) Several genetic alterations corrupt the microenvironment to better support FL. Mutations in CREBBP and deletions in B2M genes reduce MHC II and MHC I expression, respectively. On the contrary, aberrant CATHEPSIN B leads to an increase in MHC II expression and CD8 expansion. Both EZH2 and RRAGC mutations reduce the need for TFH help making FL cells more dependent on FDCs, while disruption of the HVEM–BTLA axis allows uncontrolled TFH support to FL cells.
The FL–LN maintains a structure reminiscent of a normal LN, where B cells are supported by TFH and the follicles are delimitated for a network of FDCs. These types of dendritic cells are only present in the follicles of primary and secondary lymph organs. FDCs are particular Ag-presenting cells (APCs) as they do not internalize, process, and present Ag, but present intact Ag–Ab complexes on their cell surface that induce survival of FL cells and their differentiation into memory B cells or plasma cells. In vitro studies using a non-immortalized FDC cell line have demonstrated that FDCs preferentially bind to GC B cells and deliver a positive signal for B cell survival, activation, and differentiation [11,12]. Interestingly, FL cells are then able to present this Ag derived from FDC presentation and trigger TFH recruitment. TFH are specialized CD4+ T cells located in the GC light zone and are characterized by CD4+, CXCR5+, PD1+, ICOS+, and CD25- phenotype. TFH are essential for the formation and maintenance of GC, contributing to B cell fitness by means of CD40L signaling and IL-4 or IL-21 cytokines [13]. It is noteworthy that IL-4 proteins are five-fold more abundant in FL germinal centers than in normal tonsil [14]. Moreover, malignant B cells are involved in the recruitment of TREG (CD4+, CD25+), present in a higher frequency in FL compared with tonsils, and acting as inhibitors of CD8+ T cell effector activity [15].
FRCs are stromal cells present in the T cell zone of the LN that are endowed with functions that create a permissive niche by secreting components of the extracellular matrix (ECM), including laminin, fibronectin, and collagen IV. They organize and regulate immune cell trafficking, differentiation, and migration of T cells through an IL-4/CXCL12 communication axis with TFH, among other signals, and secretion of additional chemokines such as CCL19 and CCL21. FRC also plays a direct role in B malignant cells’ activation and survival through BAFF signaling [16–19].
MSCs are present both in the BM niche and LNs, supporting B cell survival through the secretion of numerous factors, such as BAFF, TNFα, lymphotoxin α (LTα) [20], while chemokine CCL2 favors the recruitment of macrophages to the FL niche [21].
Tumor-associated macrophages (TAMs) are highly plastic cells from the myeloid lineage. Depending on the stimuli, macrophages can be polarized into M1 (inflammatory phenotype) or M2 (anti-inflammatory), resulting in distinct cytokine production or T cell function (Th1 and Th2). We have recently demonstrated that FL-FDC niche promotes, via the secretion of CCL2 and CSF-1, monocyte recruitment, differentiation, and polarization towards an M2-like pro-tumoral phenotype, as seen in FL patient biopsies [5], favoring angiogenesis, dissemination, and immunosuppression [22]. Moreover, macrophages express C-type lectin dendritic cell–specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) that binds to mannosylated BCR, activating B cell survival independently of Ag [23–25]. In addition, monocyte/macrophages trans-present IL-15 to B cells in the FL niche and cooperate with T-cell-derived CD40L to promote IL-15-dependent B-cell proliferation [26].
Thus, FL is surrounded by a rich and well-interconnected network of supportive allies that may account for the incurability of this indolent lymphoma. Moreover, this dynamic microenvironment also takes part in the histological transformation (HT) of FL to an aggressive lymphoma. These modifications comprise the disruption of the FDC network [27], changes in the gene expression of CD4/8 T cells leading to decrease motility [28], a decrease in the number and follicular distribution of FoxP3+ TREG [29] and PD-1 positive T cells. [30]
2. FL Mutational Landscape and Microenvironment Interplay
Although t(14;18) (q32;q21) was discovered decades ago [31], it is considered the first oncogenic hit in FL. It is not present in approximately 10% of patients [32,33], and it has been detected in some healthy individuals [34,35]. Therefore, additional mutations must contribute to disease onset. In this genomic era, the FL genome has been fully characterized by whole genome and exome sequencing and, more recently, by single-cell RNA sequencing (sc-RNAseq). In this review, we focus on those mutations with an impact on the FL microenvironment remodeling.
FL is a malignancy addicted to epigenetic mutations [36] where these hits (KTMD2, CREBBP/EP300, and EZH2 genes) constitute early oncogenic events present in virtually all FL patients [37–39]. These genes are involved in the post-translational modification of histones [40]. The loss of function of histone K3K4 methyltransferase KMT2D (also known as MLL2) is the most frequent alteration (60–90%), followed by loss of function mutations in the H3K27/H3K18 acetyltransferases CREBBP (50–70%) and EP300 (10–20%), resulting in transcriptional repression [41,42].
Histone-lysine N-methyltransferase 2D (KMT2D) function is related to GC formation and CSR, two critical steps in the maturation process of B cells, and it cooperates with BCL-2 in lymphomagenesis [41,43]. No impact on the immune microenvironment has been described thus far.
CREB-binding protein (CREBBP) is a haploinsufficient tumor suppressor that acts as a major regulator of enhancer networks in the GC, especially in the light zone, and avoids terminal B cell differentiation [42]. CREBBP also acetylates non-histone proteins such as p53 and BCL6. Loss of function CREBBP mutations leads to reduced activation of p53 as well as to diminished inactivation of BCL6 [44,45]. Noteworthy, CREBBP mutations have a clear effect on the immune microenvironment. MHC class II is reduced in FL cells at both the transcriptome and protein levels, resulting in a diminished Ag presentation [46]. Recently, this finding has been confirmed by scRNA-seq analysis [47]. Furthermore, CREBBP mutations are associated with reduced T cell proliferation and have been identified as early mutations, since they are present in the earliest inferable progenitors [46] and could even be present in hematopoietic stem and progenitor cell compartments [45]. Altogether, there is a large amount of evidence indicating that CREBBP mutations may play a major role in the evasion of immune surveillance during the development of FL tumors. In addition, both mutCREBBP and mutEP300 contribute to lymphomagenesis by enabling unopposed suppression of enhancers by BCL6/SMRT/HDAC3 complexes, suggesting HDAC3-targeted therapy as a precision approach for CREBBP-mutant lymphomas [48], and recent results with specific HDAC3 inhibitors have demonstrated the reactivation of immune responses [49]. While specific HDAC3 inhibitors are not at a clinical stage, diminished chromatin acetylation by mutCREBBP might be reverted using pan-HDACi. Preclinical data suggest that these families of compounds may be beneficial in combination with immunotherapy in B cell lymphomas [50]. Some clinical trials have explored pan-HDAC in monotherapy. Vorinostat yielded moderate responses (<50%) in two phase II clinical trials [51,52], while abexinostat has shown an improved overall response rate (ORR) [53]. The lack of isoform-specificity could lead to immunosuppressive effects by pan-HDACi [54].
This entry is adapted from the peer-reviewed paper 10.3390/cancers13040641