IRES is associated with obesity, glucose intolerance, dyslipidemia, and hypertension, evolves toward type 2 diabetes, and increases the risk of developing cardiovascular diseases. Several studies designed to explore the mechanisms involved in IRES allowed the identification of a multitude of potential molecular targets. Among the most promising, G Protein Coupled Receptor Kinase type 2 (GRK2) appears to be a suitable one given its functional implications in many cellular processes. In this review, we will discuss the metabolic role of GRK2 in those conditions that are characterized by insulin resistance (diabetes, hypertension, heart failure), and the potentiality of its inhibition as a therapeutic strategy to revert both insulin resistance and its associated phenotypes
- GRK2
- insulin resistance
- signaling
- adrenergic receptor
- mitochondria
1. Introduction
Insulin is a peptide hormone produced in the pancreas by the β cells of the Langerhans islets in response to the increase in plasma glucose levels. It induces the rapid absorption of glucose, protein, and fatty acids by tissues for metabolism, storage, and energy production[1] (Figure 1). Alterations in insulin signaling and production, as observed in insulin resistance, significantly impair glucose homeostasis in several pathological conditions, such as diabetes, hypertension, and heart failure. G Protein Coupled Receptor Kinase type 2 (GRK2) has been identified as a mechanism of impaired glucose homeostasis in insulin resistance and its complications. In this review, we will discuss the metabolic role of GRK2 in insulin resistance related conditions.
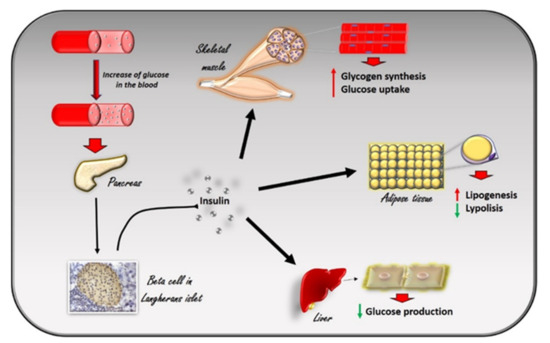
Figure 1. The effects of insulin in response to the increase in glucose levels in the blood. The increase in glucose in the blood stimulates beta cells in the pancreas to produce insulin. Circulating insulin exerts several effects in different tissues. In skeletal muscle, it promotes glucose utilization and storage by increasing glucose transport and glycogen synthesis. In the liver, insulin promotes glycogen synthesis, and inhibits glycogenolysis, gluconeogenesis, and ketogenesis. In white adipocyte tissue, insulin promotes the deposition of triglycerides, inhibits lipolysis, and promotes the absorption of glucose and fatty acids.
2. Insulin Signaling and the Development of Insulin Resistance
Glucose homeostasis is the result of a balance between glucose uptake in organs in the fed state (glycogen synthesis and glucose metabolism) and production of glucose by the liver during fasting (glycogenolysis and gluconeogenesis) and is under the control of a constellation of hormones, from insulin to glucagon, IGF-1, leptin, adiponectin, and adrenergic mechanisms. Insulin is the most potent regulator in the liver, muscle, and adipocytes. Insulin inhibits hepatic glucose output and enhances glucose uptake into skeletal muscle and adipose tissue. The insulin insensitive glucose transporter GLUT-2 in the liver and the insulin-sensitive transporter GLUT-4 in muscle and fat remove glucose from the bloodstream[2]. In muscle and adipose tissue, insulin acts by increasing the number of GLUT 4 at the plasma membrane to induce glucose transport[3][4]. In the liver, the number of GLUT-2 does not change in response to insulin, which in turn regulates the enzymes needed for maintaining glucose cellular disposal[4]. At the molecular level, insulin activates a specific tyrosine kinase receptor, the insulin receptor (IR). Insulin binding to its receptor activates its intrinsic tyrosine kinase activity (Figure 2). IR phosphorylates different substrate adaptors (IRS) in tyrosine residues that form binding sites for intracellular molecules that contain Src-homology 2 (SH2) domains[5][6]. IRS proteins are a family of adaptor proteins whose main role is to convert the tyrosine phosphorylation signal into a lipid kinase signal[5][6]. PI3K is the main active mediator of insulin effects, via the activation of the AKT/PKB cascades. Activated AKT induces glycogen synthesis through inhibition of GSK-3, protein synthesis via mTOR signaling, cell survival through inhibition of several pro-apoptotic agents, and autophagy by inhibiting FoxO transcription factors. AKT also regulates the translocation of the insulin-sensitive glucose transporter GLUT-4 to the cell membrane within muscle and fat cells for the extraction of glucose [5]. Insulin also acts through the mitogen-activated protein kinase (MAPK) pathway, by phosphorylating the IRS proteins, Gab1, and Shc [7]. The activation of the MAPK cascade is associated with the proliferative effects of insulin[7][8].
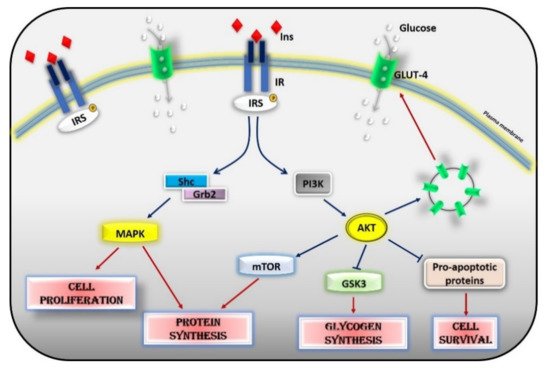
Figure 2. Insulin signaling cascade. Insulin activates the insulin receptor, which phosphorylates IRS in tyrosine residues. IRS, in turn, activates MAPK via SHc/Grb2 and AKT via PI3K. Activated AKT induces glycogen synthesis through inhibition of GSK-3, protein synthesis via mTOR signaling, cell survival through inhibition of several pro-apoptotic agents, and the translocation of GLUT-4 to the cell membrane. Activated MAPK induces cell proliferation and protein synthesis. Abbreviations: INS = Insulin; IR = Insulin Receptor; IRS = Insulin Receptor Substrate; Shc = Src Homology 2 Domain-Containing; Grb2 = Growth factor Receptor-Bound protein 2; MAPK = Mitogen-Activated Protein Kinase; PI3K = Phosphoinositide 3-kinase; AKT = Protein kinase B; mTOR = Mammalian Target of Rapamycin; GSK3 = Glycogen synthase kinase 3; GLUT4 = Glucose Transporter type 4.
Phosphorylation of serine residues in IRS1 could exert both positive and negative effects on insulin signaling[9]. Data from previous studies show a time-dependent regulation of phosphorylative events leading to early activation and inhibition of the insulin pathway through IRS-1. Positive regulatory sites are early phosphorylated to activate the insulin signaling pathway. On the contrary, the inhibitory sites are phosphorylated later to turn off the insulin signaling and disrupt the interaction between IRS1 and IR, and to promote IRS1 degradation[10]. Insulin signaling is therefore strictly dependent on a correct balance between the phosphorylation of positive and negative sites, which is altered in pathophysiological conditions. Insulin resistance (IRES) occurs when the excess of glucose in the blood induces an increase in insulin release that makes the target organs resistant to its action. Insulin resistance is a hallmark of two pathological conditions: metabolic syndrome and type 2 diabetes. Indeed, it is associated with obesity, glucose intolerance, dyslipidemia, and hypertension, which can successively progress to type 2 diabetes and cardiovascular diseases. It also increases the risk of cancer[11]. The precise pathogenetic mechanism of IRES is not yet clear but several molecular mechanisms (oxidative stress, inflammation, insulin receptor mutation, ER stress, mitochondrial dysfunction) have been demonstrated to be involved[10][12][13]. Chronic inflammation in targeted tissues was shown to contribute to altering the metabolic state, both locally via autocrine/paracrine cytokine signaling and systemically via endocrine cytokine signaling [14]. Reactive oxygen species (ROS) cause insulin resistance in the peripheral tissues by decreasing the expression of GLUT4 in the plasma membrane[15]. Then, mitochondrial alteration and obesity contribute to propagating oxidative stress [15]. In the skeletal muscle, mitochondrial dysfunction causes the development of insulin resistance in different ways: (1) by activating several protein kinases, such as Protein kinase C (PKC) or c-Jun N-terminal kinase (JNK), which inhibit IRS-1 signaling by phosphorylation; (2) by reducing the expression levels of IRS-1[16]; and (3) by inducing ER stress through excessive cytosolic calcium levels[17]. Several factors can influence insulin sensitivity such as obesity and fat distribution[18][19], age and sex[19], genetics, exercise[20], dietary nutrients[21], and hormones (glucagon, epinephrine, cortisol, leptin, growth hormones, sex hormones, amylin). Obesity is associated with an increased risk of developing insulin resistance. Indeed, the production of many mediators of glucose metabolism by the adipose tissue (Non Esterified Fatty Acids, glycerol, leptin, and adiponectin; proinflammatory cytokines) are increased in obesity[22]. In addition, mutations in genes involved in the insulin signaling cascade (insulin receptor, PI3-kinase and AKT) cause severe insulin resistance [2][23] as well as mutations and polimorphisms in genes that predispose to obesity (melanocortin-4 receptor, leptin receptor, peroxisome proliferator-activated receptor PPARg).
Insulin signaling is very complex and involves a large number of molecules that are all crucial to activate insulin-dependent biological processes. Therefore, several events could regulate insulin effects and sensitivity, such as protein degradation and synthesis, phosphorylative events, post-translational modifications, as well as the inhibition of single components at different steps of the long insulin-dependent cascade. In this context, the G Protein-Coupled Receptor Kinase 2 (GRK2) is considered a key regulator of metabolic processes and, in particular, of glucose metabolism.
3. GRK2 Affects Insulin Signaling
G Protein-Coupled Receptor Kinases (GRK) are known to phosphorylate G Protein Coupled Receptors GPCRs on the plasma membrane to turn off their intracellular signaling[24][25]. Therefore, GRKs exert a critical role in both physiological states and pathological conditions that are mainly associated with alterations of the adrenergic signaling. Besides this classical effect, GRKs are also known to inactivate non-GPCR receptors, including tyrosine kinase receptors, by phosphorylative events[24]. Moreover, they can interact with and regulate the activity of several cytosolic substrates[26][27][28]. Therefore, GRKs are involved in the regulation of key processes within the cell and are, consequently, potential therapeutic targets [26][29]. Among GRKs, GRK2 is known to regulate cell metabolism[29][30][31]. GRK2 shuttles among the different subcellular compartments following the energetic requests of the cell[32][33]. In particular, we showed that GRK2 localizes in mitochondria and regulates critical mitochondrial processes, such as ATP production and mitochondrial biogenesis [32]. The overexpression of the kinase favors such processes preventing ATP loss after hypoxia/reperfusion[32] and mitochondrial damage in response to acute exposure to ionizing radiation[34]. Despite energy metabolism, the suggestion of GRK2 involvement in cell metabolism comes from the demonstration that insulin induces an up-regulation of the kinase expression levels and the association between GRK2 and IRS1[35]. GRK2 phosphorylates IRS1 in serine/threonine and inhibits the activation of the downstream signaling. In particular, GRK2 inhibits the phosphorylation of IRS1 in Tyr612, blocking the insulin signaling, and induces its phosphorylation in Ser307, promoting IRS degradation[35][36]. These results are in line with the reciprocal relationship between these two sites of IRS phosphorylation that is described in previous reports[37][38]. Chronic stimulation of beta-adrenergic receptors (βAR) is involved in the pathogenesis of insulin resistance. In cells overexpressing the βAR, GRK2 accumulates in the plasma membrane and inhibits IRS1 activation and glucose uptake in response to insulin. The selective inhibition of the kinase potentiates insulin signaling both in vitro and in vivo suggesting that GRK2 mediates adrenergic insulin resistance while its inhibition increases insulin sensitivity [35]. These observations point to GRK2 as a potential target, also considering that conditions associated with insulin resistance (diabetes, hypertension, or chronic activation of β adrenergic receptor), are all characterized by elevated kinase levels. Indeed, GRK2 expression is increased in key tissues in different experimental models of insulin resistance, and its downregulation ameliorates the alterations of glucose homeostasis and insulin signaling in response to different insults[39]. Recently, the role of GRK2 in myeloid cells has also been investigated. Vila-Bedmar and colleagues suggest that the downregulation of GRK2 in these cells reduces the pro-inflammatory features of macrophages and prevents high fat diet-induced metabolic alterations [40]. GRK2 expression levels, as well as its subcellular localization and activity, are finely regulated. Several molecules are involved in regulating different steps of kinase transcription, mRNA regulation and translation, protein maturation, localization, activation, and degradation, as extensively described in the review from Penela and colleagues[41]. Here, we will discuss the recent findings on the metabolic role of GRK2 in insulin resistance-related conditions, such as diabetes, hypertension, and heart failure.
This entry is adapted from the peer-reviewed paper 10.3390/cells10010167
References
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223.
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814.
- Dugani, C.B.; Klip, A. Glucose transporter 4: Cycling, compartments and controversies. Embo. Rep. 2005, 6, 1137–1142.
- Chadt, A.; Al-Hasani, H. Glucose transporters in adipose tissue, liver, and skeletal muscle in metabolic health and disease. Pflug. Arch. 2020, 472, 1273–1298.
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44.
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191, doi:10.1101/cshperspect.a009191.
- Kong, L.; Wang, Q.; Jin, J.; Xiang, Z.; Chen, T.; Shen, S.; Wang, H.; Gao, Q.; Wang, Y. Insulin resistance enhances the mito-gen-activated protein kinase signaling pathway in ovarian granulosa cells. PLoS ONE 2017, 12, e0188029.
- Biddinger, S.B.; Kahn, C.R. From mice to men: Insights into the insulin resistance syndromes. Annu. Rev. Physiol. 2006, 68, 123–158.
- Weigert, C.; Kron, M.; Kalbacher, H.; Pohl, A.K.; Runge, H.; Haring, H.U.; Schleicher, E.; Lehmann, R. Interplay and effects of temporal changes in the phosphorylation state of serine-302, -307, and -318 of insulin receptor substrate-1 on insulin ac-tion in skeletal muscle cells. Mol. Endocrinol. 2008, 22, 2729–2740.
- Tanti, J.F.; Jager, J. Cellular mechanisms of insulin resistance: Role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr Opin Pharm. 2009, 9, 753–762.
- Vigneri, R.; Goldfine, I.D.; Frittitta, L. Insulin, insulin receptors, and cancer. J. Endocrinol. Investig. 2016, 39, 1365–1376.
- Sesti, G. Pathophysiology of insulin resistance. Best Pr. Res. Clin. Endocrinol Metab 2006, 20, 665–679.
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell Physiol. 2019, 234, 8152–8161.
- de Luca, C.; Olefsky, J.M. Inflammation and insulin resistance. Febs. Lett. 2008, 582, 97–105.
- Hurrle, S.; Hsu, W.H. The etiology of oxidative stress in insulin resistance. Biomed. J. 2017, 40, 257–262.
- Ryu, H.S.; Park, S.Y.; Ma, D.; Zhang, J.; Lee, W. The induction of microRNA targeting IRS-1 is involved in the development of insulin resistance under conditions of mitochondrial dysfunction in hepatocytes. PLoS ONE 2011, 6, e17343.
- Lim, J.H.; Lee, H.J.; Ho Jung, M.; Song, J. Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: A molecular mechanism leading to hepatic insulin resistance. Cell Signal. 2009, 21, 169–177.
- Kahn, S.E.; Prigeon, R.L.; Schwartz, R.S.; Fujimoto, W.Y.; Knopp, R.H.; Brunzell, J.D.; Porte, D. Jr. Obesity, body fat distribu-tion, insulin sensitivity and Islet beta-cell function as explanations for metabolic diversity. J. Nutr. 2001, 131, 354S–560S.
- Karakelides, H.; Irving, B.A.; Short, K.R.; O'Brien, P.; Nair, K.S. Age, obesity, and sex effects on insulin sensitivity and skel-etal muscle mitochondrial function. Diabetes 2010, 59, 89–97.
- Borghouts, L.B.; Keizer, H.A. Exercise and insulin sensitivity: A review. Int J. Sports Med. 2000, 21, 1–12.
- Galgani, J.E.; Uauy, R.D.; Aguirre, C.A.; Diaz, E.O. Effect of the dietary fat quality on insulin sensitivity. Br. J. Nutr. 2008, 100, 471–479.
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846.
- Parker, V.E.; Savage, D.B.; O'Rahilly, S.; Semple, R.K. Mechanistic insights into insulin resistance in the genetic era. Diabet. Med. 2011, 28, 1476–1486.
- Ribas, C.; Penela, P.; Murga, C.; Salcedo, A.; Garcia-Hoz, C.; Jurado-Pueyo, M.; Aymerich, I.; Mayor, F. Jr. The G pro-tein-coupled receptor kinase (GRK) interactome: Role of GRKs in GPCR regulation and signaling. Biochim. Biophys. Acta 2007, 1768, 913–922.
- Gambardella, J.; Sorriento, D.; Bova, M.; Rusciano, M.; Loffredo, S.; Wang, X.; Petraroli, A.; Carucci, L.; Mormile, I.; Oliveti, M.; et al. Role of Endothelial G Protein-Coupled Receptor Kinase 2 in Angioedema. Hypertension 2020, 76, 1625–1636.
- Penela, P.; Murga, C.; Ribas, C.; Lafarga, V.; Mayor, F. Jr. The complex G protein-coupled receptor kinase 2 (GRK2) interac-tome unveils new physiopathological targets. Br. J. Pharm. 2010, 160, 821–832.
- Sorriento, D.; Ciccarelli, M.; Santulli, G.; Campanile, A.; Altobelli, G.G.; Cimini, V.; Galasso, G.; Astone, D.; Piscione, F.; Pastore, L.; et al. The G-protein-coupled receptor kinase 5 inhibits NFkappaB transcriptional activity by inducing nuclear accumulation of IkappaB alpha. Proc. Natl. Acad. Sci. USA 2008, 105, 17818–17823.
- Sorriento, D.; Santulli, G.; Ciccarelli, M.; Maione, A.S.; Illario, M.; Trimarco, B.; Iaccarino, G. The Amino-Terminal Domain of GRK5 Inhibits Cardiac Hypertrophy through the Regulation of Calcium-Calmodulin Dependent Transcription Factors. Int. J. Mol. Sci. 2018, 19, 861, doi:10.3390/ijms19030861.
- Sorriento, D.; Gambardella, J.; Fiordelisi, A.; Iaccarino, G.; Illario, M. GRKs and beta-Arrestins: "Gatekeepers" of Mitochon-drial Function in the Failing Heart. Front. Pharm. 2019, 10, 64.
- Ciccarelli, M.; Cipolletta, E.; Iaccarino, G. GRK2 at the control shaft of cellular metabolism. Curr. Pharm. Des. 2012, 18, 121–127.
- Sorriento, D.; Ciccarelli, M.; Santulli, G.; Illario, M.; Trimarco, B.; Iaccarino, G. Trafficking GRK2: Cellular and Metabolic consequences of GRK2 subcellular localization. Transl. Med. Unisa. 2014, 10, 3–7.
- Fusco, A.; Santulli, G.; Sorriento, D.; Cipolletta, E.; Garbi, C.; Dorn, G.W. 2nd; Trimarco, B.; Feliciello, A.; Iaccarino, G. Mitochondrial localization unveils a novel role for GRK2 in organelle biogenesis. Cell Signal. 2012, 24, 468–475.
- Sorriento, D.; Fusco, A.; Ciccarelli, M.; Rungi, A.; Anastasio, A.; Carillo, A.; Dorn, G.W. 2nd; Trimarco, B.; Iaccarino, G. Mi-tochondrial G protein coupled receptor kinase 2 regulates proinflammatory responses in macrophages. Febs. Lett. 2013, 587, 3487–3494.
- Franco, A.; Sorriento, D.; Gambardella, J.; Pacelli, R.; Prevete, N.; Procaccini, C.; Matarese, G.; Trimarco, B.; Iaccarino, G.; Ciccarelli, M. GRK2 moderates the acute mitochondrial damage to ionizing radiation exposure by promoting mitochondrial fission/fusion. Cell Death Discov. 2018, 4, 25.
- Cipolletta, E.; Campanile, A.; Santulli, G.; Sanzari, E.; Leosco, D.; Campiglia, P.; Trimarco, B.; Iaccarino, G. The G protein coupled receptor kinase 2 plays an essential role in beta-adrenergic receptor-induced insulin resistance. Cardiovasc. Res. 2009, 84, 407–415.
- Ciccarelli, M.; Chuprun, J.K.; Rengo, G.; Gao, E.; Wei, Z.; Peroutka, R.J.; Gold, J.I.; Gumpert, A.; Chen, M.; Otis, N.J.; et al. G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia. Circulation 2011, 123, 1953–1962.
- Shahid, G.; Hussain, T. GRK2 negatively regulates glycogen synthesis in mouse liver FL83B cells. J. Biol. Chem. 2007, 282, 20612–20620.
- Aguirre, V.; Werner, E.D.; Giraud, J.; Lee, Y.H.; Shoelson, S.E.; White, M.F. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J. Biol. Chem. 2002, 277, 1531–1537.
- Garcia-Guerra, L.; Nieto-Vazquez, I.; Vila-Bedmar, R.; Jurado-Pueyo, M.; Zalba, G.; Diez, J.; Murga, C.; Fernandez-Veledo, S.; Mayor, F. Jr.; Lorenzo, M. G protein-coupled receptor kinase 2 plays a relevant role in insulin resistance and obesity. Diabe-tes 2010, 59, 2407–2417.
- Vila-Bedmar, R.; Cruces-Sande, M.; Arcones, A.C.; Willemen, H.; Prieto, P.; Moreno-Indias, I.; Diaz-Rodriguez, D.; Francisco, S.; Jaen, R.I.; Gutierrez-Repiso, C.; et al. GRK2 levels in myeloid cells modulate adipose-liver crosstalk in high fat di-et-induced obesity. Cell Mol. Life Sci. 2020, 77, 4957–4976.
- Penela, P.; Ribas, C.; Sanchez-Madrid, F.; Mayor, F. Jr. G protein-coupled receptor kinase 2 (GRK2) as a multifunctional sig-naling hub. Cell Mol. Life Sci. 2019, 76, 4423–4446.