PCOS as the most common endocrine disorder of women in their reproductive age affects between 5–15% of the female population. Apart from its cardinal symptoms, like irregular and anovulatory cycles, hyperandrogenemia and a typical ultrasound feature of the ovary, obesity, and insulin resistance are often associated with the disease. Furthermore, PCOS represents a status of chronic inflammation with permanently elevated levels of inflammatory markers including IL-6 and IL-18, TNF-α, and CRP. Inflammation, as discovered only recently, consists of two processes occurring concomitantly: active initiation, involving “classical” mediators including prostaglandins and leukotrienes, and active resolution processes based on the action of so-called specialized pro-resolving mediators (SPMs). These novel lipid mediator molecules derive from the essential ω3-poly-unsaturated fatty acids (PUFAs) DHA and EPA and are synthesized via specific intermediates. The role and benefits of SPMs in chronic inflammatory diseases like obesity, atherosclerosis, and Diabetes mellitus has become a subject of intense research during the last years and since PCOS features several of these pathologies, this review aims at summarizing potential roles of SPMs in this disease and their putative use as novel therapeutics.
- PCOS
- obesity
- inflammation
1. Introduction
Polycystic ovary syndrome (PCOS) is a wide-spread endocrine disorder affecting 5–15% of women in their reproductive age worldwide and is a frequent cause of infertility[1]. Diagnosis according to current international guidelines that apply the diagnostic “Rotterdam” criteria require the presence of at least two of the following features: hyperandrogenism, ovulatory dysfunction or/and polycystic ovary in ultrasonic scans, while other pathologies must be excluded[2]. PCOS is further characterized by an elevated LH/FSH ratio and often associated with obesity and insulin resistance leading to an hyperinsulinemic state in 80% of the obese women and 30–40% of the lean ones [3]. The etiology of the syndrome has not been clarified completely yet. Certain genetic patterns that impact for example synthesis, regulation and action of sex hormones and the insulin receptor, but also gestational factors like high maternal levels of androgens or AMH (anti-Müllerian hormone) seem to contribute to the onset of the disease[4][5][6]. Postnatal lifestyle factors like inadequate nutrition accompanied by a lack of physical exercise promote the development of the disease as they often result in obesity and disturbances of the glucose metabolism. In fact, hyperinsulinemia, independent of BMI, is a key contributor to PCOS pathogenesis[7][8] as it results in augmented androgen production in the adrenal cortex and follicles via stimulation of LH secretion, while concomitantly reducing SHBG (sex hormone binding globulin) synthesis[8]. Resulting elevated androgene levels interfere with follicular maturation[9] and may lead to the characteristic clinical manifestations like acne and hirsutism. Obesity on the other hand, may not only reinforce insulin resistance[10], but is a pathogenicity factor itself contributing to the endocrinologic disorder due to the altered hormone metabolism of adipocytes[11]. Furthermore, adipose tissue contributes to constant low-level systemic inflammation as will be described further below in this review. PCOS itself nowadays is considered a condition of chronic inflammation with elevated levels of leukocytes, pro-inflammatory cytokines, elevated white blood count and markers such as the C-reactive protein being detectable [12] and also affects women with a normal BMI[13].
2. Inflammation: Initiation and Active Resolution
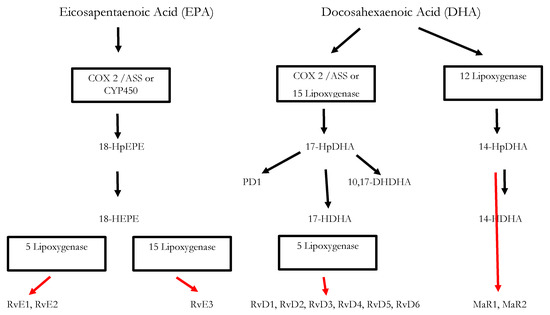
Inflammatory processes are crucial for the survival of human beings and occur as a reaction to stimuli like injury or infections. Potential pathogens may enter a host due to a trauma, barrier breakage or microbial invasion and in order to regain the integrity of the organism, the invaders must be eliminated, removed and the functional state of affected tissue must be restored. During the acute inflammatory response, eicosanoid lipid mediator (LM) molecules that derive from the ω-6 poly-unsaturated fatty acid (PUFA) arachidonic acid (AA) are rapidly synthesized by cells of the innate immune system, such as granulocytes and macrophages that are recruited to the sited of the event[14]. For this step, the enzymes cyclooxagenases 1 and 2 (Cox-1/2) are utilized and prostanoids including prostaglandins (PG), leukotrienes (LTs) and thromboxanes (TXs) are synthesized from AA[15][16]. Additionally, mast cells and the classically activated M1 macrophages secrete different kinds of inflammatory cytokines including TNFα, IL-1 and Il-6—all of them being highly inflammatory substances that drive the generation of the classical inflammatory symptoms: redness, heat, swelling, pain, and loss of function[14][15][16]. Guided by the inflammatory eicosanoids and cytokines present at the site of infection, neutrophiles and monocytes leave the blood vessels and migrate into the tissue, thereby contributing to the formation of inflammatory exudates and progression of the inflammation process[14][15][16]. An effective initiation of the inflammatory response is essential for survival, but its self-limitation is equally important. Exceeding inflammation may lead to the phenomenon of a cytokine storm and subsequent life-threatening sepsis [17]. The failure to temporally limit inflammation results in chronic inflammatory diseases including cardiovascular and neurological disorders, auto-immune diseases, diabetes, and cancer[18] [19]. For a long time, the resolution of inflammation had been considered a passive process with mere dissolution of inflammatory mediators and inflammation divided into initiation and resolution[20]. This view was changed by the discovery of distinct lipid mediator molecules that can “switch on” resolution processes in animal models and actively drive the resolution processes. These mediators comprise four different subgroups: the resolvins (Rvs), lipoxins (LXs), protectins (PDs) and maresins (Masr) and are depicted as SPMs: specialized pro-resolving mediators[21][22][23]. Resolvins, protectins, and maresins are synthesized from the ω-3 PUFAs EPA (Eicosapentanenoic acid) and DHA (docosahexaenoic acid) via certain intermediates (18-HpETE, 17-HpDHA and 14-HpDHA). Their biosynthetic pathways (see Figure 1 for an overview) involve certain lipoxygenases as well as the Cox-enzymes that take part both in the eicosanoid synthesis as well as in SPM production. The synthesis of LX in contrast starts from ω-6 PUFA AA[24][25]. Interestingly, Aspirin as an irreversible inhibitor of Cox-enzymes blocks prostanoid synthesis by modification of their catalytic domain. However, these enzymes remain active and are triggered to synthesize the SPM precursors 18-HpETE and 17-HpDHA. Resulting SPMS are called aspirin-triggered resolvins, -maresins, or protectins (AT-SPMs) and are potent mediators of resolution that are widely used in experimental approaches[21][24][26][27].
3. Significance of SPMs in the Resolution of Inflammatory Processes
4. Chronic Inflammatory Diseases: Significance of SPMs
Insufficient resolution may result in chronic inflammation and SPMs play a crucial role in its suppression as was deduced from experiments with different animal models[24][35] The tissue damage observed in Periodontitis, for example, can be attributed to the action of activated neutrophils that produce pro-inflammatory cytokines PGE2, LTB4 and LXA4. An infection with the causative oral pathogen P. gingivalis results in a strong inflammatory response leading to an upregulation of Cox-2, and recruitment of neutrophiles as demonstrated in Air Pouche mouse models. Supplementation of lipoxin LXA4 analogues was shown to reduce both processes. Within these mouse models also the development of systemic inflammation due to an infection with the oral pathogen has been demonstrated, as Cox-2 expression in further tissues like lung and heart is triggered by the infection. Utilizing a rabbit animal model that overexpresses lipoxygenase type I leading to LX4-levels that are up to 10 times higher than in wild type rabbits further demonstrated the importance of the SPM LX4 in this disease: Periodontitis-provoked bone-loss was reduced in transgenic animals and neutrophil infiltration was significantly reduced compared to wild type animals[35].
In a further experimental approach with transgenic mice, the role of 12/15 lipoxygenase and its biosynthesis products including SPMs LXA4, RvD1 and RvD2 for atherosclerosis was demonstrated. Atherosclerosis is initiated by an inflammatory reaction of the vascular endothelial cells that, vial endothelial secretion of pro-inflammatory cytokines, lead to recruitment of monocytes and leukocytes. Monocytes differentiate into macrophages that transform into foam cells clustered at the vessel walls. Resulting atheroma enlarge and narrow the vessel. Foam cells inside the plaques continue to secrete pro-inflammatory cytokines, reactive oxygen species and other mediators. Atherosclerosis is therefore considered a chronic inflammatory disease[36]. Interestingly, transgenic 12/15 lipoxygenase (12/15 LOX) mice seemed to be protected against atherosclerosis and this was attributed to the elevated expression levels of RvD1, PD1 and 17-HpDHA compared to wild type mice. It was further shown that LXA4, PD1 and RvD1 reduced the number of cytokines derived from endothelial cells as well as the amount of adhesion molecules (P-Selectin and VCAM-1) and additionally improved the uptake of apoptotic thymocytes. All these processes contributed to the anti-atherogenic influence supporting the notion that atherosclerosis is a result of vascular non-resolving inflammation. Interestingly, in these transgenic 12/15 LOX mice, a standard high-fat western diet disrupted the protective mechanisms[37] and proved the impact of nutrition on inflammation homeostasis. Further evidence for the resolutive action of SPMs in chronic inflammatory diseases were derived from a murine model of arthritis, in which resolvin RvD1 and its metabolic precursor 17-HpDHA were shown to reduce tissue damage and pain more efficiently than steroids[38].
The clinical picture of fibrosis may result from unresolved inflammation and epithelial or microvascular bruises. In animal models the role of exogenous aspirin-triggered LX4-analogues in the reduction of pulmonal fibrosis was demonstrated [39] and in further experiments both LXA4 and its analogue benzo-LXA4 were shown to reduce fibrosis in kidney[39][40][41]. Further trials have demonstrated that exogenously administered RvD1 can reduce the amount of pro-inflammatory mediator molecules that are formed after exposure to cigarette smoke and lung toxins [42] and a reduction in SPM levels has been associated the chronic lung diseases asthma and COPD in humans [43].
6. Chronic Inflammation in PCOS
In most cases PCOS is accompanied by obesity and insulin resistance (IR), affecting about 65-80% of all patients [44][45] and it is well-known that hyperinsulinemia, hyperandrogenism and obesity in this disease reinforce each other[8][11][45][46]. But PCOS also represents a state of chronic inflammation[47] resulting in part from excess visceral adipose tissue and its above-described pro-inflammatory mechanisms. Chronic low-level inflammation mirrored by elevated levels of pro-inflammatory cytokines, however, is also present in normal-weight PCOS-affected women but was mainly attributed to the fact that also normal-weight PCOS patients tend to have a surplus of visceral adipose tissue and intraperitoneal fat depots[48][49]. Recent studies have identified BMI and insulin resistance as main predictors for increased levels of CRP and white blood cells[12]. In addition, PCOS patients were shown to have a certain pro-inflammatory genotype characterized by alterations in the genes encoding for TNF-α, TNF receptor and IL-6 [50][51][52]. Furthermore, hyperandrogenaemia in PCOS not only contributes to enhanced visceral adiposity but was also shown to contribute to inflammatory processes. Excess androgens trigger the activation of MNC (mononuclear cells) which leads to enhanced production of reactive oxygen species (ROS) and activation of NFκB, which in turn, enhances the expression of the pro-inflammatory cytokines TNF-α, IL-6, and Il-1. TNF-α and IL-6 are known mediators of insulin resistance and hyperandrogenaemia was therefore found to have a negative impact on the insulin-mediated IRS-PI3K-Akt signaling pathway[53][54].
These correlations represent an important linkage of the metabolic features in PCOS and chronic inflammation. Furthermore, abnormal ovarian function has been associated with enhanced macrophage infiltration of the ovary and increased expression of TNF-α, IL-6 and IL-8 resulting in the activation of corresponding pro-inflammatory signaling pathways [55][56][57].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22010384
References
- Azziz, R.; Carmina, E.; Chen, Z.; Dunaif, A.; Laven, J.S.E.; Legro, R.S.; Lizneva, D.; Horowtiz-Natterson, B.; Teede, H.J.; Yildiz, B.O. Polycystic ovary syndrome. Nat. Rev. Dis. Primers 2016; 2, 16057, doi:10.1038/nrdp.2016.57.
- European Society of Human Reproduction and Embryology. International Evidence-Based Guideline for the Assessment and Management of Polycystic Ovary Syndrome. 2018. Available at: https://www.eshre.eu/Guidelines-and-Legal/Guidelines/Polycystic-Ovary-Syndrome (accessed on 28 October 2020).
- Ehrmann, D.A.; Barnes, R.B.; Rosenfield, R.L.; Cavaghan, M.K.; Imperial, J. Prevalence of impaired glucose tolerance and diabetes in women with polycystic ovary syndrome. Diabetes Care 1999, 22, 141–146, doi:10.2337/diacare.22.1.141. PMID: 10333916.
- Diamanti-Kandarakis, E.; Kandarakis, H.; Legro, R.S. The Role of Genes and Environment in the Etiology of PCOS. Endocrine 2006, 30, 19–26, doi:10.1385/endo:30:1:19.
- De Melo, A.S.; Dias, S.V.; Cavalli, R.C.; Cardoso, V.C.; Bettiol, H.; Barbieri, M.A.; Ferriani, R.A.; Vieira, C.S.; De Melo, A.S. Pathogenesis of polycystic ovary syndrome: Multifactorial assessment from the foetal stage to menopause. Reproduction 2015, 150, R11–R24, doi:10.1530/rep-14-0499.
- Daghestani, M.H. Rs1799817 in INSR associates with susceptibility to polycystic ovary syndrome. J. Med Biochem. 2019, 39, 149–159, doi:10.2478/jomb-2019-0023.
- Ciampelli, M.; Fulghesu, A.; Cucinelli, F.; Pavone, V.; Ronsisvalle, E.; Guido, M.; Caruso, A.; Lanzone, A. Impact of insulin and body mass index on metabolic and endocrine variables in polycystic ovary syndrome. Metabolism 1999, 48, 167–172, doi:10.1016/s0026-0495(99)90028-8.
- Diamanti-Kandarakis, E.; Dunaif, A. Insulin resistance and the polycystic ovary syndrome revisited: An update on mechanisms and implications. Endocr. Rev. 2012, 33, 981–1030, doi:10.1210/er.2011-1034.
- Dewailly, D.; Robin, G.; Peigne, M.; Decanter, C.; Pigny, P.; Catteau-Jonard, S. Interactions between androgens, FSH, anti-Müllerian hormone and estradiol during folliculogenesis in the human normal and polycystic ovary. Hum. Reprod. Update 2016, 22, 709–724, doi:10.1093/humupd/dmw027.
- Kahn, S.E.; Hull, R.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006; 444, 840–846, doi:10.1038/nature05482.
- Rojas, J.; Chávez, M.; Olivar, L.; Rojas, M.; Morillo, J.; Mejías, J.; Calvo, M.; Bermúdez, V. Polycystic ovary syndrome, insulin resistance, and obesity: Navigating the pathophysiologic labyrinth. Int. J. Reprod. Med. 2014, 2014, 1–17, doi:10.1155/2014/719050.
- Rudnicka, E.; Kunicki, M.; Suchta, K.; Machura, P.; Grymowicz, M.; Smolarczyk, R. Inflammatory markers in women with polycystic ovary syndrome. BioMed Res. Int. 2020, 2020, 4092470, doi:10.1155/2020/4092470.
- Chazenbalk, G.; Chen, Y.H.; Heneidi, S.; Lee, J.M.; Pall, M.; Chen, Y.D.I.; Azziz, R. Abnormal expression of genes involved in inflammation, lipid metabolism, and Wnt signaling in the adipose tissue of polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2012, 97, E765–70, doi:10.1210/jc.2011-2377.
- Chiurchiù, V.; Leuti, A.; Maccarrone, M. Bioactive lipids and chronic inflammation: Managing the fire within. Front. Immunol. 2018, 9, 38, doi:10.3389/fimmu.2018.00038.
- Flower, R.J. Prostaglandins, bioassay and inflammation. Br. J. Pharmacol. 2006, 147, S182–S192, doi:10.1038/sj.bjp.0706506.
- Samuelsson, B. Role of basic science in the development of new medicines: Examples from the Eicosanoid field. J. Biol. Chem. 2012, 287, 10070–10080, doi:10.1074/jbc.x112.351437.
- Hotchkiss, R.R.; Moldawer, L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.I.; Vincent, J.-L. Sepsis and septic shock. Nat. Rev. Dis. Prim. 2016, 2, 1–21, doi:10.1038/nrdp.2016.45.
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832, doi:10.1038/s41591-019-0675-0.
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882, doi:10.1016/j.cell.2010.02.029.
- Tabas, I.; Glass, C.K. Anti-inflammatory therapy in chronic disease: Challenges and opportunities. Science 2013, 339, 166–172, doi:10.1126/science.1230720.
- Serhan, C.N.; Clish, C.B.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel Functional sets of lipid-derived mediators with antiinflammatory actions generated from Omega-3 fatty acids via cyclooxygenase 2–nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 2000, 192, 1197–1204, doi:10.1084/jem.192.8.1197.
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nat. Cell Biol. 2014, 510, 92–101, doi:10.1038/nature13479.
- Serhan, C.N.; Levy, B.D. Resolvins in inflammation: Emergence of the pro-resolving superfamily of mediators. J. Clin. Investig. 2018, 128, 2657–2669, doi:10.1172/jci97943.
- Serhan, C.N.; Chiang, N.; Dalli, J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin. Immunol. 2015, 27, 200–215, doi:10.1016/j.smim.2015.03.004.
- Serhan, C.N. Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J. 2017, 31, 1273–1288, doi:10.1096/fj.201601222r.
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.L. Resolvins. J. Exp. Med. 2002, 196, 1025–1037, doi:10.1084/jem.20020760.
- Serhan, C.N.; Fredman, G.; Yang, R.; Karamnov, S.; Belayev, L.S.; Bazan, N.G.; Zhu, M.; Winkler, J.W.; Petasis, N.A. Novel proresolving aspirin-triggered DHA pathway. Chem. Biol. 2011, 18, 976–987, doi:10.1016/j.chembiol.2011.06.008.
- Valor-Méndez, L.; Neurath, M.F. Resolution of chronic inflammatory disease: universal and tissue-specific concepts. Nat. Commun. 2018, 9, 1–8, doi:10.1038/s41467-018-05800-6.
- Serhan, C.N.; Chiang, N. Resolution phase lipid mediators of inflammation: agonists of resolution. Curr. Opin. Pharmacol. 2013, 13, 632–640, doi:10.1016/j.coph.2013.05.012.
- Serhan, C.N.; Savill, J. Resolution of inflammation: the beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197, doi:10.1038/ni1276.
- Bandeira-Melo, C.; Serra, M.F.; Diaz, B.L.; Cordeiro, R.S.B.; Silva, P.M.R.; Lenzi, H.L.; Bakhle, Y.S.; Serhan, C.N.; Martins, M.A. Cyclooxygenase-2-derived prostaglandin E2 and lipoxin A4 accelerate resolution of allergic edema in Angiostrongylus costaricensis-infected rats: Relationship with concurrent Eosinophilia. J. Immunol. 2000, 164, 1029–1036, doi:10.4049/jimmunol.164.2.1029.
- Levy, B.D.; Clish, C.B.; A Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: signals in resolution. Nat. Immunol. 2001, 2, 612–619, doi:10.1038/89759.
- Barnig, C.; Cernadas, M.; Dutile, S.; Liu, X.; Perrella, M.A.; Kazani, S.; Wechsler, M.E.; Israel, E.; Levy, B.D. Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci. Transl. Med. 2013, 5, 174ra26, doi:10.1126/scitranslmed.3004812.
- Chiurchiù, V.; Leuti, A.; Dalli, J.; Jacobsson, A.; Battistini, L.; Maccarrone, M.; Serhan, C.N. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci. Transl. Med. 2016, 8, 353ra111, doi:10.1126/scitranslmed.aaf7483.
- Serhan, C.N.; Jain, A.; Marleau, S.; Clish, C.; Kantarci, A.; Behbehani, B.; Colgan, S.P.; Stahl, G.L.; Merched, A.; Petasis, N.A.; et al. Reduced inflammation and tissue damage in transgenic rabbits overexpressing 15-lipoxygenase and endogenous anti-inflammatory lipid mediators. J. Immunol. 2003, 171, 6856–6865, doi:10.4049/jimmunol.171.12.6856.
- Merched, A.J.; Ko, K.; Gotlinger, K.H.; Serhan, C.N.; Chan, L. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 2008, 22, 3595–3606, doi:10.1096/fj.08-112201.
- Merched, A.J.; Serhan, C.N.; Chan, L. Nutrigenetic disruption of inflammation-resolution homeostasis and atherogenesis. J. Nutr. Nutr. 2011, 4, 12–24, doi:10.1159/000326890.
- Lima-Garcia, J.F.; Dutra, R.C.; Da Silva, K.; Motta, E.M.; Campos, M.M.; Calixto, J.B. The precursor of resolvin D series and aspirin-triggered resolvin D1 display anti-hyperalgesic properties in adjuvant-induced arthritis in rats. Br. J. Pharmacol. 2011, 164, 278–293, doi:10.1111/j.1476-5381.2011.01345.x.
- Martins, V.; Valenca, S.S.; Farias-Filho, F.A.; Molinaro, R.; Simões, R.L.; Ferreira, T.P.T.; E Silva, P.M.R.; Hogaboam, C.M.; Kunkel, S.L.; Fierro, I.M.; et al. ATLa, an aspirin-triggered lipoxin A4 synthetic analog, prevents the inflammatory and fibrotic effects of bleomycin-induced pulmonary fibrosis. J. Immunol. 2009, 182, 5374–5381, doi:10.4049/jimmunol.0802259.
- Börgeson, E.; Docherty, N.G.; Murphy, M.; Rodgers, K.; Ryan, A.; O’Sullivan, T.P.; Guiry, P.J.; Goldschmeding, R.; Higgins, D.F.; Godson, C. Lipoxin A 4 and benzo‐lipoxin A 4 attenuate experimental renal fibrosis. FASEB J. 2011, 25, 2967–2979, doi:10.1096/fj.11-185017
- Qu, X.; Zhang, X.; Yao, J.; Song, J.; Nikolic-Paterson, D.J.; Li, J. Resolvins E1 and D1 inhibit interstitial fibrosis in the obstructed kidney via inhibition of local fibroblast proliferation. J. Pathol. 2012, 228, 506–519, doi:10.1002/path.4050.
- Hsiao, H.-M.; Sapinoro, R.E.; Thatcher, T.H.; Croasdell, A.; Levy, E.P.; Fulton, R.A.; Olsen, K.C.; Pollock, S.J.; Serhan, C.N.; Phipps, R.P.; et al. A novel anti-inflammatory and pro-resolving role for Resolvin D1 in acute cigarette smoke-induced lung inflammation. PLOS ONE 2013, 8, e58258, doi:10.1371/journal.pone.0058258.
- Duvall, M.G.; Levy, B.D. DHA- and EPA-derived resolvins, protectins, and maresins in airway inflammation. Eur. J. Pharmacol. 2016, 785, 144–155, doi:10.1016/j.ejphar.2015.11.001.
- Marshall, J.C.; Dunaif, A. Should all women with PCOS be treated for insulin resistance? Fertil. Steril. 2012, 97, 18–22, doi:10.1016/j.fertnstert.2011.11.036.
- Al-Jefout, M.; Alnawaiseh, N.; Al-Qtaitat, A. Insulin resistance and obesity among infertile women with different polycystic ovary syndrome phenotypes. Sci. Rep. 2017, 7, 5339, doi:10.1038/s41598-017-05717-y.
- Aytan, A.N.; Bastu, E.; Demiral, I.; Bulut, H.; Dogan, M.; Buyru, F. Relationship between hyperandrogenism, obesity, inflammation and polycystic ovary syndrome. Gynecol. Endocrinol. 2016, 32, 709–713, doi:10.3109/09513590.2016.1155208.
- Patel, S. Polycystic ovary syndrome (PCOS), an inflammatory, systemic, lifestyle endocrinopathy. J. Steroid Biochem. Mol. Biol. 2018, 182, 27–36.
- Carmina, E.; Bucchieri, S.; Esposito, A.; Del Puente, A.; Mansueto, P.; Orio, F.; Di Fede, G.; Rini, G. Abdominal fat quantity and distribution in women with polycystic ovary syndrome and extent of its relation to insulin resistance. J. Clin. Endocrinol. Metab. 2007, 92, 2500–2505, doi:10.1210/jc.2006-2725.
- Gonzalez, F.; Thusu, K.; Abdel-Rahman, E.; Prabhala, A.; Tomani, M.; Dandona, P. Elevated serum levels of tumor necrosis factor alpha in normal-weight women with polycystic ovary syndrome. Metabolism 1999, 48, 437–441, doi:10.1016/s0026-0495(99)90100-2.
- Escobar-Morreale, H.F.; Calvo, R.M.; Villuendas, G.; Sancho, J.; Millán, J.L.S. Association of polymorphisms in the Interleukin 6 receptor complex with obesity and hyperandrogenism. Obes. Res. 2003, 11, 987–996, doi:10.1038/oby.2003.136.
- González, F. Inflammation in polycystic ovary syndrome: Underpinning of insulin resistance and ovarian dysfunction. Steroids 2012, 77, 300–305, doi:10.1016/j.steroids.2011.12.003.
- Talaat, R.M.; Mohamed, Y.A.; Mohamad, E.H.; Elsharkawy, M.; Guirgis, A. Interleukin 10 (− 1082 G/A) and (− 819 C/T) gene polymorphisms in Egyptian women with polycystic ovary syndrome (PCOS). Meta Gene 2016, 9, 254–258, doi:10.1016/j.mgene.2016.08.001.
- González F, Rote NS, Minium J, Kirwan JP, Minium, J. Insulin sensitivity and hyperandrogenism in polycystic ovary syndrome are related to activated nuclear factor kB from mononuclear cells in the fasting state. In Proceedings of the 89th Meeting of the Endocrine Society, Toronto, Canada, 2–5 June 2007; pp. 142.
- Zhang, Y.; Meng, F.; Sun, X.; Sun, X.; Hu, M.; Cui, P.; Vestin, E.; Li, X.; Li, W.; Wu, X.-K.; et al. Hyperandrogenism and insulin resistance contribute to hepatic steatosis and inflammation in female rat liver. Oncotarget 2018, 9, 18180–18197, doi:10.18632/oncotarget.24477.
- Xie, F.; Anderson, C.L.; Timme, K.R.; Kurz, S.G.; Fernando, S.C.; Wood, J.R. Obesity-dependent increases in oocyte mRNAs are associated with increases in proinflammatory signaling and gut microbial abundance of Lachnospiraceae in female mice. Endocrinology 2016, 157, 1630–1643, doi:10.1210/en.2015-1851.
- Nteeba, J.; Ortinau, L.; Perfield, J.; Keating, A.F. Diet-induced obesity alters immune cell infiltration and expression of inflammatory cytokine genes in mouse ovarian and peri-ovarian adipose depot tissues. Mol. Reprod. Dev. 2013, 80, 948–958, doi:10.1002/mrd.22231.
- Skaznik-Wikiel, M.E.; Swindle, D.C.; Allshouse, A.A.; Polotsky, A.J.; McManaman, J.L. High-fat diet Causes subfertility and compromised ovarian function independent of obesity in mice1. Biol. Reprod. 2016, 94, 108, doi:10.1095/biolreprod.115.137414.