Cryptococcosis is an important opportunistic infection and a leading cause of meningitis in patients with HIV infection. The current antifungal pharmacological treatment is limited; in addition, the high toxicity, increased resistance rate, and difficulty of the currently available antifungal molecules to cross the blood-brain barrier hamper the treatment. Thus, the search for new alternatives for the treatment of cryptococcosis is extremely necessary. Here we describe the therapeutic strategies currently available and discuss new molecules with antifungal potential in different phases of clinical trials and in the advanced pre-clinical phase.
- Cryptococcus
- antifungal
- synthetic molecules
- Drug Repurposing
- Immunobiological
Current Therapy
The treatment of cryptococcal meningitis consists of three phases: induction (2 weeks), consolidation (8 weeks) and maintenance (6–12 months). The guidelines of the Society for Infectious Diseases of America [1] and the World Health Organization [2] emphasize the importance of the use of potent fungicidal drugs during the induction phase; however, worldwide access to antifungal drugs is still inadequate [3], which highlights the importance of alternative treatment strategies.
The primary therapy of cryptococcal meningitis depends on the condition of the patients infected with Cryptococcus. For HIV-infected, HIV-non infected and non-transplanted individuals, the primary therapy consists on the induction with AMB (0.7–1.0 mg/kg/day) plus 5-FC (100 mg/kg/day) for 2 weeks. For consolidation and maintenance, FLC at 400 mg/day for 8 weeks (minimum) and at 200 mg/day for 6–12 months, respectively, are employed. In addition, there are other alternative regimens; for example, in case of AMB intolerance, LAMB (3–4 mg/kg/day) or ABLC (5 mg/kg/day) can be used. If 5-FC is not used, AMB deoxycholate or AMB lipid formulations should be maintained for at least 2 weeks [1].
For patients with nonmeningeal cryptococcosis forms as pulmonary (immunosuppressed and nonimmunosupressed) and nonpulmonary cryptococcosis, FLC (400 mg/day) for 6–12 months is recommended. For the pulmonary (nonimmunosupressed) form, voriconazole (VRC) (200 mg twice/day), itraconazole (ITC) (200 mg/day), and posaconazole (POS) (400 mg twice/day) are acceptable alternatives if FLC is unavailable or contraindicated [1].
Amphotericin B (AMB)
Despite AMB dose-limiting toxicity, it has remained the gold standard for treating disseminated life-threatening fungal infections [4]. Its fungicidal effect is associated with AMB binding to ergosterol in the membranes of fungal cell (Figure 1) [5][6]. AMB perturbs the membrane function, causing leakage of cellular contents, and leads to death by cellular dysfunction [5][6]. The first commercially available formulation was Fungizone®, a conventional micellar form of AMB and deoxycholate. Currently, parenteral formulations based on lipid carriers are also available, and they include a liposomal formulation (LAMB), a lipid complex formulation (ABLC), and a colloidal dispersion (ABCD) [7]. Their main advantage is the reduction of side effects of AMB [8].
Resistance to AMB is rare and often caused by a decrease in the amount of ergosterol in the plasma membrane or a change in the target sterol, which leads to a decrease in the binding of AMB [1][9]. Some fungal cells have a mutation in the ergosterol biosynthesis pathway, producing ergosterol-like compounds instead of ergosterol, which have lower binding affinity for AMB [10][11].
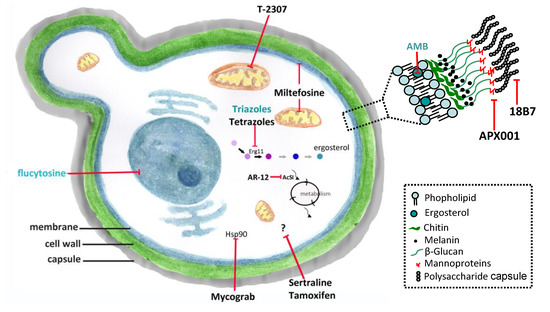
Figure 1. Conventional antifungals and new molecules for cryptococcosis treatment. Amphotericin B (AMB) and azoles inhibit ergosterol and its biosynthesis, respectively, and flucytosine inhibits the nucleic acids synthesis. New molecules acting on non-conventional targets or different structures of fungal cells may have antifungal effects. Erg11 (or Cyp51)—cytochrome P450-dependent lanosterol C14-alpha-demethylase; AcS—Acetyl CoA synthetase; Hsp90—Heat shock protein 90. (previously published in the review DOI:10.3390/
Flucytosine (5-FC)
5-FC was synthesized in 1957 as a potent antitumoral agent [12][13]. 5-FC is taken into the fungal cell by cytosine permease, and its action as an antifungal agent depends on its conversion to 5-fluorouracil (5-FU) within of the target cells. 5-FU becomes incorporated to the RNA and inhibits DNA synthesis by thymidylate synthase inhibition (Figure 1). It is most active agent against yeasts, including Candida and Cryptococcus spp. [14]; however, the occurrence of resistance to 5-FC prevents its use as a single agent [14][15][16][17][18]. Currently, its use is indicated only in combination with other antifungals, mainly AMB [8][14]. 5-FC exhibits significant adverse effects, in particular hepatotoxicity and myelotoxicity, which is probably due to toxic 5-FU plasma concentrations.
Fluconazole (FLC)
FLC is a triazole agent that inhibits the fungal cytochrome P450-dependent lanosterol C14-alpha-demethylase (Erg11 or Cyp51) leading to ergosterol biosynthesis inhibition (Figure 1) [19][20][21]. FLC diffuses easily into the cerebrospinal fluid, sputum and saliva, and is concentrated in the urine and skin [22]. The most frequent adverse effects are gastrointestinal events, headache and skin rashes; isolated instances of clinically evident hepatic dysfunction have occurred in patients with AIDS [23]. Over the years, there has been a gradual increase of resistance to FLC in clinical isolates of C. neoformans, and nowadays, resistance is a relatively common event in relapse episodes of cryptococcal meningitis [19]. FLC resistance phenotype in Cryptococcus spp. have been associated with mutations in the ERG11 gene [19][24][25]. However, heteroresistance in Cryptococcus spp. can lead to higher FLC tolerance by selection of heteroresistant clones after induction due to previous exposure to FLC [19][26].
Voriconazole (VRC)
VRC was developed to increase the antifungal spectrum of available triazoles. To reach this goal, the molecule of FLC was modified, with the substitution of the fluoropyrimidine ring for one of the azole groups, and addition of the α-methyl group to provide fungicidal activity against molds [27][28]. The most frequently reported adverse effect of VRC is transient visual disturbances, that are often associated with higher doses, and considerable hepatotoxic effects. In addition, studies reported important drug interactions with VRC [27]. An ongoing clinical trial study (from 2020 to 2022), named “Three Induction Treatments on Cryptococcal Meningitis (TITOC)”, is investigating its use for cryptococcosis treatment at the Hospital of the University of Zhejiang, China (NCT04072640, www.clinicaltrials.gov). Resistance to this azole is not a common event, but there are reports in the literature in recent years [29][30][31][32].
New Molecules and Drug Repurposing
Immunobiological and new molecules acting on non-conventional targets or other structures of the fungal cell might have potential as antifungal agents. In this context, drug repositioning is an interesting strategy for antifungal discovery because pharmacokinetics and safety data in humans have been previously established. Therefore, expanding the application of a drug to additional diseases is both cost and time-effective [33][34]. In this section, we will discuss new molecules and drugs currently used in the treatment of other diseases that have activity against Cryptococcus spp. (Figure 1).
Interferon-Gamma (IFN-γ)
IFN-γ is an endogenous cytokine with several biological properties and activities, including a key role in the host response to intracellular pathogens, directing the immune system towards the protective Th1 type immunity [35]. Exogenous IFN-γ has been investigated as a potential adjunct agent for treatment of cryptococcal meningitis. In a murine model of pulmonary and disseminated infection, IFN-γ administration resulted in the decrease of the fungal burden in the infected organs, and significantly extended mice survival [36]. One phase II clinical trial (NCT00012467) suggested that IFN-γ may provide rapid and early sterilization of CNS in patients with HIV-associated cryptococcal meningitis without pronounced adverse effects [35]. However, in another study, it was observed that even though administration of IFN-γ improved the fungal clearance from the CNS, it failed to significantly decrease patient mortality [37].
Mycograb
Mycograb is a recombinant human antibody against fungal heat shock protein 90 (Hsp90), which are chaperones required for the maintenance of cellular homeostasis in various fungal pathogens [38][39]. Cryptococcus neoformans isolates were susceptible to mycograb at MIC values from 256 to 1024 µg/mL, and a synergistic effect was observed in combination with AMB [40]. The efficacy and safety of mycograb for cryptococcal meningitis are under evaluation in ongoing phase II clinical trials (NCT00324025 and NCT00847678).
18B7
18B7 is a monoclonal antibody directed against the capsular polysaccharide of C. neoformans. Administration of 18B7 promoted rapid clearance of serum cryptococcal antigen and deposition in the liver and spleen, and presented no reactivity with normal mouse, rat, or human tissues [41]. It also reduced the fungal burden in tissues, improved granuloma formation, and demonstrated synergism with AMB, FLC and 5-FC in mice [42][43][44][45]. 18B7 was evaluated in a phase I clinical trial in HIV-infected patients with cryptococcal meningitis, being well tolerated in doses up to 1 mg/kg without evidence of toxicity [46].
APX001 (Fosmanogepix)/APX001A (Manogepix)
APX001 (prodrug of APX001A) is a first-in-class small-molecule antifungal drug candidate that inhibits the fungal enzyme Gwt1 (an inositol acylase) in the glycosylphosphatidylinositol (GPI) biosynthesis pathway [47]. The APX001A MIC ranged from 0.03 to 2 µg/mL for 48 Cryptococcus spp. clinical isolates in vitro [48][49], and demonstrated in vitro synergism with FLC [49]. APX001 alone or in combination with FLC decreased the fungal burden in the lungs and brain using cryptococcal meningitis murine model [49]. Other structural analogues of APX001A also demonstrated an excellent in vitro inhibitory effect on C. neoformans; and using in vivo assay APX2096 (prodrug of APX2039) led to a nearly complete or complete sterilization of lungs and brain [49]. The preclinical efficacy of APX001/APX001A against Cryptococcus associated with previous safety and pharmacological data (NCT02957929 and NCT02956499) lend support to further clinical evaluation of the molecule for treatment of human cryptococcosis.
T-2307
T-2307 is a novel arylamidine derivative with broad-spectrum of action and potent in vitro and in vivo activities, that acts by selectively disrupting mitochondrial function in yeasts [50]. The antifungal activity for C. neoformans was observed at MIC ranging from 0.0039 to 0.0625 µg/mL [51], and for C. gattii at 0.0078–0.0625 µg/mL [52]. The efficacy of T-2307 was confirmed in murine models of cryptococcosis: at 0.1 mg/kg, T-2307 significantly delayed mortality in mice infected by C. neoformans when compared with the untreated group, and T-2307 exhibited a superior protective effect compared to AMB at similar treatment regimens [51]. Administration of T-2307 alone at 2 mg/kg/day significantly reduced viable cell counts in the lungs and brain of mice infected by C. gattii and the results were similar to standard treatments [52].
Sertraline
Sertraline is an antidepressant that belongs to the group of selective inhibitors of serotonin reuptake. Initially used for treatment of major depressive disorder, it is now also approved for management of obsessive-compulsive, panic and post-traumatic stress disorders [53]. Although its mechanism of action on fungi has not fully elucidated, inhibition of protein synthesis in Cryptococcus spp. has been described [54]. In vitro studies showed that sertraline is effective to inhibit Cryptococcus growth at 1–8 µg/mL; in contrast to FLC, sertraline showed fungicidal effect at concentrations higher than 6 µg/mL [54][55]. Murine cryptococcosis model confirmed the antifungal activity observed in vitro, in which sertraline at 15 mg/kg decreased the fungal burden in the brain and spleen when compared with the untreated group [54][55]. Sertraline combined with FLC in vitro showed either additive or synergistic effects, and in animal models, this drug combination led to fungal clearance at a greater rate than either drug alone [54][56][57]. Sertraline use for cryptococcal meningitis treatment alone or in combination with AMB and FLC was investigated in phase III clinical trials (NCT01802385 and NCT03002012), and these studies demonstrated that sertraline did not reduce the mortality rate of patients. This lack of efficacy appears to be multifactorial, and might be associated with insufficient duration of therapeutic sertraline concentrations [58].
Tamoxifen
Tamoxifen belongs to the pharmacological class of selective estrogen receptor modulators; it is an estrogen receptor agonist in the bone, cardiovascular system, and endometrium, while acting as an estrogen receptor antagonist in the breast tissue. This drug is clinically used to treat and prevent breast cancer and osteoporosis [59]. Tamoxifen has in vitro antifungal activity against Cryptococcus spp. clinical isolates, with MIC ranging from 2 to 16 µg/mL, acting synergistically when combined with AMB and FLC [60][61]. In the murine disseminated cryptococcosis model, treatment with tamoxifen at 200 mg/kg/day combined with FLC at 5 mg/kg/day decreased the burden fungal by ~1 log in the brain tissue [61]. The authors of the study suggested the use of this drug for treatment of cryptococcosis because high concentrations (well above of the MIC values) were reached in the CNS in addition to the antifungal activity inside macrophages, synergism with existing therapies AMB and FLC, and good oral bioavailability [59][61]. At the moment, clinical trials (phase II) are being carried out to evaluate the efficacy, feasibility, and safety of tamoxifen in combination with standard therapies (AMB and FLC) in the treatment of cryptococcal meningitis (NCT03112031). Although tamoxifen activity against Cryptococcus has been reported, and the drug is under evaluation in ongoing clinical trials for cryptococcosis treatment, its mechanism of action has not been elucidated yet.
AR-12
AR-12, a small molecule derived from celecoxib, was tested as an antitumoral agent in phase I clinical trials, and licensed to Arno Therapeutics (NCT00978523) [62]. AR-12 is a non-nucleoside acetyl CoA synthetase inhibitor as previously investigated in S. cerevisiae and C. albicans [63]. This molecule has broad-spectrum antifungal activity, including for C. neoformans, with MIC value of 4 µg/mL, and AR-12 was demonstrated to be effective in a murine model of disseminated cryptococcosis when combined with FLC (dose at 100 and 10 mg/kg, respectively), decreasing the fungal burden in the brain [64].
Miltefosine (MFS)
MFS belongs to the alkylphosphocholine class of molecules, and is used in the treatment of cutaneous metastases of breast cancer and leishmaniasis [65]. Studies showed that MFS has a broad-spectrum in vitro antifungal activity, including against C. gattii and C. neoformans isolates in the both planktonic (0.25–4 µg/mL) and biofilm (8 - ≥16 µg/mL) lifestyles [66][67][68]. Moreover, MFS was effective to control the fungal infection in the larval model of Galleria mellonella by C. gattii at 10, 20, and 40 mg/kg [69]. MFS at 3.6 and 7.2 mg/kg/day has shown effectiveness in the murine model of disseminated cryptococcosis [67] although this result has been conflicting with other work [70]. This contradiction demonstrates variable translation of in vitro MFS activity to in vivo murine models of disseminated cryptococcosis. Studies evidenced that MFS acts through multiple mechanisms, being able to alter membrane permeability, inhibit phospholipase B1 and induce an apoptotic-like cell death reducing the mitochondrial membrane potential, increasing reactive oxygen species (ROS) production, and inducing DNA fragmentation and condensation [67][68]. Despite the extensive and exciting in vitro reports highlighting MFS usefulness as an antifungal drug, no clinical trial for treatment of fungal infections is under way.
Tetrazoles
Tetrazoles are synthetic molecules produced from azoles, non-metabolized bioisosteric analogs of carboxylic acid and cis-amide; they possess diverse chemotherapeutic properties and are highly selective fungal Cyp51 inhibitor [71][72]. Among tetrazoles, VT-1129 and VT-1598 are more selective for fungal Cyp51 than mammalian Cyp450 enzymes and both molecules showed antifungal efficacy against Cryptococcus spp. [73][74]. VT-1129 inhibited the growth of C. neoformans and C. gattii isolates at 0.003–4 µg/mL and 0.06–8 µg/mL, respectively [75][76]. VT-1598 has lower MIC values (0.06 to 0.15 µg/mL) [77][78]. In addition to the in vitro models, assays using cryptococcosis murine models demonstrated that oral administration of VT-1598 resulted in suitable plasma and brain concentrations, leading to a significant reduction in the brain fungal burden [78]. Recently, phase I clinical trials have started to assess the safety and pharmacokinetics of VT-1598 (NCT04208321).
This entry is adapted from the peer-reviewed paper 10.3390/microorganisms8040613
References
- John R. Perfect; William E. Dismukes; Francoise Dromer; David L. Goldman; John R. Graybill; Richard J. Hamill; Thomas S Harrison; Robert A. Larsen; Olivier Lortholary; Minh‐Hong Nguyen; et al. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the infectious diseases society of america. Clinical Infectious Diseases 2010, 50, 291-322, 10.1086/649858.
- World Health Organization. Guidelines for the Diagnosis, Prevention and Management of Cryptococcal Disease in HIV-Infected Adults, Adolescents and Children: Supplement to the 2016 Consolidated Guidelines on the Use of Antiretroviral Drugs for Treating and Preventing HIV Infection; World Health Organization: Geneva, Switzerland, 2018; pp. 1–51.
- John R. Perfect; Tihana Bicanic; Cryptococcosis diagnosis and treatment: What do we know now. Fungal Genetics and Biology 2014, 78, 49-54, 10.1016/j.fgb.2014.10.003.
- David Ellis; Amphotericin B: spectrum and resistance. Journal of Antimicrobial Chemotherapy 2002, 49, 7-10, 10.1093/jac/49.suppl_1.7.
- Brandon C. Wilcock; Matthew M. Endo; Brice E. Uno; Martin D. Burke; C2′-OH of Amphotericin B Plays an Important Role in Binding the Primary Sterol of Human Cells but Not Yeast Cells. Journal of the American Chemical Society 2013, 135, 8488-8491, 10.1021/ja403255s.
- Min Liu; Meiwan Chen; Zhiwen Yang; Design of amphotericin B oral formulation for antifungal therapy. Drug Delivery 2017, 24, 1-9, 10.1080/10717544.2016.1225852.
- Cuddihy, G.; Wasan, E.K.; Di, Y.; Wasan, K.M. The development of oral amphotericin B to treat systemic fungal and parasitic infections: Has the myth been finally realized? Pharmaceutics 2019, 11, 99.
- Romuald Bellmann; Piotr Smuszkiewicz; Pharmacokinetics of antifungal drugs: practical implications for optimized treatment of patients. Infection 2017, 45, 737-779, 10.1007/s15010-017-1042-z.
- Lozano-Chiu, M.; Rex, J.H. Resistance to antifungal agents. In Topley & Wilson’s Microbiology and Microbial Infections, 9th ed.; Arnold: London, UK, 1998; pp. 177–187.
- Wilfried Posch; Michael Blatzer; Doris Wilflingseder; Cornelia Lass-Flörl; Aspergillus terreus: Novel lessons learned on amphotericin B resistance. Medical Mycology 2018, 56, S73-S82, 10.1093/mmy/myx119.
- O’Shaughnessy, E.M.; Lyman, C.A.; Walsh, T.J. Amphotericin B: Polyene resistance mechanisms. In Antimicrobial Drug Resistance; Humana Press: Totowa, NJ, USA, 2009; pp. 295–305.
- Duschinsky, R.; Pleven, E.; Heidelberger, C; The synthesis of 5-Fluoropyrimidines. J. Am. Chem. Soc. 1957, 79, 4559–4560, .
- A. Vermes; Henk-Jan Guchelaar; J. Dankert; Flucytosine: a review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. Journal of Antimicrobial Chemotherapy 2000, 46, 171-179, 10.1093/jac/46.2.171.
- Vermes, A.; Guchelaar, H.; Dankert, J. Flucytosine: A review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J. Antimicrob. Chemother. 2000, 46, 171–179.
- Tassel, D.; Madoff, M. Treatment of Candida sepsis and Cryptococcus meningitis with 5-Fluorocytosine. JAMA 1968, 206, 830.
- Polak, A.; Scholer, H.J. Mode of action of 5-Fluorocytosine and mechanisms of resistance. Chemotherapy 1975, 21, 113–130.
- Normark, S.; Schonebeck, J. In Vitro studies of 5-Fluorocytosine resistance in Candida albicans and Torulopsis glabrata. Antimicrob. Agents Chemother. 1972, 2, 114–121.
- Pfaller, M.A. Antifungal drug resistance: Mechanisms, epidemiology, and consequences for treatment. Am. J. Med. 2012, 125, S3–S13.
- Bongomin, F.; Oladele, R.O.; Gago, S.; Moore, C.B.; Richardson, M.D. A systematic review of fluconazole resistance in clinical isolates of Cryptococcus species. Mycoses 2018, 61, 290–297.
- Richardson, K. The discovery and profile of Fluconazole. J. Chemother. 1990, 2, 51–54.
- Kartalija, M.; Kaye, K.; Tureen, J.H.; Liu, Q.; Tauber, M.G.; Elliott, B.R.; Sande, M.A. Treatment of experimental Cryptococcal meningitis with Fluconazole: Impact of dose and addition of Flucytosine on mycologic and pathophysiologic outcome. J. Infect. Dis. 1996, 173, 1216–1221.
- Brammer, K.W.; Farrow, P.R.; Faulkner, J.K; Pharmacokinetics and tissue penetration of Fluconazole in humans. Clin. Infect. Dis. 1990, 12, S318–S326, .
- Goa, K.L.; Barradell, L.B. Fluconazole. Drugs 1995, 50, 658–690.
- Gago, S.; Serrano, C.; Alastruey-Izquierdo, A.; Cuesta, I.; Martín-Mazuelos, E.; Aller, A.I.; Gómez-López, A.; Mellado, E. Molecular identification, antifungal resistance and virulence of Cryptococcus neoformans and Cryptococcus deneoformans isolated in Seville, Spain. Mycoses 2017, 60, 40–50.
- Rodero, L.; Mellado, E.; Rodriguez, A.C.; Salve, A.; Guelfand, L.; Cahn, P.; Cuenca-Estrella, M.; Davel, G.; Rodriguez-Tudela, J.L. G484S amino acid substitution in Lanosterol 14-α Demethylase (ERG11) is related to Fluconazole resistance in a recurrent Cryptococcus neoformans clinical isolate. Antimicrob. Agents Chemother. 2003, 47, 3653–3656.
- Sionov, E.; Chang, Y.C.; Garraffo, H.M.; Kwon-Chung, K.J. Heteroresistance to Fluconazole in Cryptococcus neoformans is intrinsic and associated with virulence. Antimicrob. Agents Chemother. 2009, 53, 2804–2815.
- Helen W. Boucher; Andreas H. Groll; Christine C. Chiou; Thomas J Walsh; Thomas J. Walsh; Newer Systemic Antifungal Agents. Drugs 2004, 64, 1997-2020, 10.2165/00003495-200464180-00001.
- Ursula Theuretzbacher; Franziska Ihle; Hartmut Derendorf; Hartmut Derendorf; Pharmacokinetic/Pharmacodynamic Profile of Voriconazole. Clinical Pharmacokinetics 2006, 45, 649-663, 10.2165/00003088-200645070-00002.
- P. Mondon; R. Petter; G. Amalfitano; R. Luzzati; E. Concia; I. Polacheck; K. J. Kwon-Chung; Heteroresistance to Fluconazole and Voriconazole inCryptococcus neoformans. Antimicrobial Agents and Chemotherapy 1999, 43, 1856-1861, 10.1128/aac.43.8.1856.
- N. Mandras; Janira Roana; Vivian Tullio; Valeria Allizond; Giuliana Banche; D. Scalas; G. Fucale; Annamaria Cuffini; A Case of Fluconazole, Voriconazole-Resistant Cryptococcus neoformans Isolated from an Immunocompetent Patient. Journal of Chemotherapy 2011, 23, 379-380, 10.1179/joc.2011.23.6.379.
- Alexander Perkins; A. Gomez-Lopez; Emilia Mellado; Juan L. Rodriguez-Tudela; Manuel Cuenca-Estrella; Rates of antifungal resistance among Spanish clinical isolates of Cryptococcus neoformans var. neoformans. Journal of Antimicrobial Chemotherapy 2005, 56, 1144-1147, 10.1093/jac/dki393.
- Rui Kano; Miki Okubo; Atsuhiko Hasegawa; Hiroshi Kamata; Multi-azole-resistant strains of Cryptococcus neoformans var. grubii isolated from a FLZ-resistant strain by culturing in medium containing voriconazole. Medical Mycology 2017, 55, 877-882, 10.1093/mmy/myw101.
- Hiroshi Tsubamoto; Tomoko Ueda; Kayo Inoue; Kazuko Sakata; Hiroaki Shibahara; Takashi Sonoda; Repurposing itraconazole as an anticancer agent. Oncology Letters 2017, 14, 1240-1246, 10.3892/ol.2017.6325.
- Megan Truong; Leigh G. Monahan; Dee A. Carter; Ian Charles; Repurposing drugs to fast-track therapeutic agents for the treatment of cryptococcosis. PeerJ 2018, 6, e4761, 10.7717/peerj.4761.
- Peter G. Pappas; Beatriz Bustamante; Eduardo Ticona; Richard J. Hamill; Philip C. Johnson; Annette Reboli; Judith Aberg; Rodrigo Hasbun; Henry H. Hsu; Recombinant Interferon‐γ1b as Adjunctive Therapy for AIDS‐Related Acute Cryptococcal Meningitis. The Journal of Infectious Diseases 2004, 189, 2185-2191, 10.1086/420829.
- Kazuyoshi Kawakami; Masaki Tohyama; Katsuji Teruya; Norifumi Kudeken; QiFeng Xie; Atsushi Saito; Contribution of interferon-γ in protecting mice during pulmonary and disseminated infection withCryptococcus neoformans. FEMS Immunology & Medical Microbiology 1996, 13, 123-130, 10.1111/j.1574-695x.1996.tb00225.x.
- Joseph N. Jarvis; Graeme Meintjes; Kevin Rebe; Gertrude Ntombomzi Williams; Tihana Bicanic; Anthony Williams; Charlotte Schutz; Linda-Gail Bekker; Robin Wood; Thomas S Harrison; et al. Adjunctive interferon-γ immunotherapy for the treatment of HIV-associated cryptococcal meningitis. AIDS 2012, 26, 1105-1113, 10.1097/qad.0b013e3283536a93.
- Ruth C Matthews; Gordon Rigg; Samantha Hodgetts; Tracey Carter; Caroline Chapman; Carl Gregory; Chris Illidge; James Burnie; Preclinical assessment of the efficacy of mycograb, a human recombinant antibody against fungal HSP90. Antimicrobial Agents and Chemotherapy 2003, 47, 2208–2216, .
- Rossana De Aguiar Cordeiro; Antonio José De Jesus Evangelista; Rosana Serpa; Francisca Jakelyne De Farias Marques; Charlline Vládia Silva De Melo; Jonathas Oliveira; Jônatas Da Silva Franco; Lucas Pereira De Alencar; Tereza De Jesus Pinheiro Gomes Bandeira; Raimunda Sâmia Nogueira Brilhante; et al. Inhibition of heat-shock protein 90 enhances the susceptibility to antifungals and reduces the virulence of Cryptococcus neoformans/Cryptococcus gattii species complex. Microbiology 2016, 162, 309-317, 10.1099/mic.0.000222.
- Lucy Nooney; Ruth C. Matthews; James P. Burnie; Evaluation of Mycograb®, amphotericin B, caspofungin, and fluconazole in combination against Cryptococcus neoformans by checkerboard and time-kill methodologies. Diagnostic Microbiology and Infectious Disease 2005, 51, 19-29, 10.1016/j.diagmicrobio.2004.08.013.
- Arturo Casadevall; Wendy Cleare; Marta Feldmesser; Aharona Glatman-Freedman; David L. Goldman; Thomas R. Kozel; Nikoletta Lendvai; Jean Mukherjee; Liise-Anne Pirofski; Johanna Rivera; et al. Characterization of a Murine Monoclonal Antibody toCryptococcus neoformans Polysaccharide That Is a Candidate for Human Therapeutic Studies. Antimicrobial Agents and Chemotherapy 1998, 42, 1437-1446, 10.1128/aac.42.6.1437.
- J. Mukherjee; M. Feldmesser; M. D. Scharff; A. Casadevall; Monoclonal antibodies to Cryptococcus neoformans glucuronoxylomannan enhance fluconazole efficacy. Antimicrobial Agents and Chemotherapy 1995, 39, 1398-1405, 10.1128/aac.39.7.1398.
- Marta Feldmesser; Jean Mukherjee; Arturo Casadevall; Combination of 5-flucytosine and capsule-binding monoclonal antibody in the treatment of murine Cryptococcus neoformans infections and in vitro. Journal of Antimicrobial Chemotherapy 1996, 37, 617-622, 10.1093/jac/37.3.617.
- F Dromer; J Charreire; Improved amphotericin B activity by a monoclonal anti-Cryptococcus neoformans antibody: study during murine cryptococcosis and mechanisms of action. The Journal of Infectious Diseases 1991, 163, 1114–1120, .
- M Feldmesser; A Casadevall; Effect of serum IgG1 to Cryptococcus neoformans glucuronoxylomannan on murine pulmonary infection. The Journal of Immunology 1997, 158, 790–799, .
- R. A. Larsen; Peter G. Pappas; John Perfect; Judith A. Aberg; Arturo Casadevall; Gretchen A. Cloud; Robert James; Scott Filler; William E. Dismukes; Phase I Evaluation of the Safety and Pharmacokinetics of Murine-Derived Anticryptococcal Antibody 18B7 in Subjects with Treated Cryptococcal Meningitis. Antimicrobial Agents and Chemotherapy 2005, 49, 952-958, 10.1128/aac.49.3.952-958.2005.
- Miao Zhao; Alexander J. Lepak; Karen Marchillo; Jamie VanHecker; Hiram Sanchez; Paul G. Ambrose; David R. Andes; APX001 Pharmacokinetic/Pharmacodynamic Target Determination against Aspergillus fumigatus in an In Vivo Model of Invasive Pulmonary Aspergillosis. Antimicrobial Agents and Chemotherapy 2019, 63, e02372-18, 10.1128/aac.02372-18.
- M. A. Pfaller; M. D. Huband; R. K. Flamm; P. A. Bien; M. Castanheira; In Vitro Activity of APX001A (Manogepix) and Comparator Agents against 1,706 Fungal Isolates Collected during an International Surveillance Program in 2017. Antimicrobial Agents and Chemotherapy 2019, 63, e00840-19, 10.1128/AAC.00840-19.
- Shaw, K.J.; Schell, W.A.; Covel, J.; Duboc, G.; Giamberardino, C.; Kapoor, M.; Moloney, M.; Soltow, Q.A.; Tenor, J.L.; Toffaletti, D.L.; et al. In Vitro and In Vivo evaluation of APX001A/APX001 and other Gwt1 inhibitors against Cryptococcus. Antimicrob. Agents Chemother. 2018, 62, e00523-18.
- Tatsuya Shibata; Toshinari Takahashi; Eio Yamada; Akiko Kimura; Hiroshi Nishikawa; Hiroyoshi Hayakawa; Nobuhiko Nomura; Junichi Mitsuyama; T-2307 Causes Collapse of Mitochondrial Membrane Potential in Yeast. Antimicrobial Agents and Chemotherapy 2012, 56, 5892-5897, 10.1128/AAC.05954-11.
- Mitsuyama, J.; Nomura, N.; Hashimoto, K.; Yamada, E.; Nishikawa, H.; Kaeriyama, M.; Kimura, A.; Todo, Y.; Narita, H; In Vitro and In Vivo antifungal activities of T-2307, a novel arylamidine. Antimicrob. Agents Chemother. 2008, 52, 1318–1324, 10.1128/AAC.05856-11.
- Hiroshi Nishikawa; Yoshiko Fukuda; Junichi Mitsuyama; Masato Tashiro; Akitaka Tanaka; Takahiro Takazono; Tomomi Saijo; Kazuko Yamamoto; Shigeki Nakamura; Yoshifumi Imamura; et al. In vitro and in vivo antifungal activities of T-2307, a novel arylamidine, against Cryptococcus gattii: an emerging fungal pathogen.. Journal of Antimicrobial Chemotherapy 2017, 72, 1709-1713, 10.1093/jac/dkx020.
- DeVane, C.L.; Liston, H.L.; Markowitz, J.S; Clinical pharmacokinetics of Sertraline. Clinical Pharmacology & Therapeutics 2002, 41, 1247–1266, .
- Bing Zhai; Cheng Wu; Linqi Wang; Matthew Sachs; Xiaorong Lin; The Antidepressant Sertraline Provides a Promising Therapeutic Option for Neurotropic Cryptococcal Infections. Antimicrobial Agents and Chemotherapy 2012, 56, 3758-3766, 10.1128/AAC.00212-12.
- Trevinõ-Rangel, R.D.J.; Villanueva-Lozano, H.; Hernández-Rodríguez, P.; Martínez-Reséndez, M.F.; Garciá-Juárez, J.; Rodríguez-Rocha, H.; González, G.M; In Vitro Antifungal Activity of Sertraline and Synergistic Effects in Combination with Antifungal Drugs against Planktonic Forms and Biofilms of Clinical Trichosporon asahii Isolates. Med. Mycol. 2016, 54, 280–286, .
- Rahul Nayak; Jianping Xu; Effects of sertraline hydrochloride and fluconazole combinations on Cryptococcus neoformans and Cryptococcus gattii. Mycology 2010, 1, 99-105, 10.1080/21501203.2010.487054.
- Michaela Spitzer; Emma Griffiths; K. M. Blakely; Jan Wildenhain; Linda Ejim; Laura Rossi; Gianfranco De Pascale; Jasna Ćurak; Eric Brown; Mike Tyers; et al. Cross‐species discovery of syncretic drug combinations that potentiate the antifungal fluconazole. Molecular Systems Biology 2011, 7, 1-14, 10.1038/msb.2011.31.
- Rhein, J.; Huppler Hullsiek, K.; Tugume, L.; Nuwagira, E.; Mpoza, E.; Evans, E.E.; Kiggundu, R.; Pastick, K.A.; Ssebambulidde, K.; Akampurira, A.; et al. Adjunctive sertraline for HIV-associated cryptococcal meningitis: a randomised, placebo-controlled, double-blind phase 3 trial. The Lancet Infectious Diseases 2019, 19, 843-851, 10.1016/S1473-3099(19)30127-6.
- Karla C. Morello; Gregory T. Wurz; Michael W. DeGregorio; Pharmacokinetics of Selective Estrogen Receptor Modulators. Clinical Pharmacokinetics 2003, 42, 361-372, 10.2165/00003088-200342040-00004.
- Trieu Phan Hai; Anh Duong Van; Nguyen Thi Thuy Ngan; Le Thanh Hoang Nhat; Nguyen Phu Huong Lan; Nguyen V. Vinh Chau; Guy E. Thwaites; Damian Krysan; J. N. Day; The combination of tamoxifen with amphotericin B, but not with fluconazole, has synergistic activity against the majority of clinical isolates of Cryptococcus neoformans. Mycoses 2019, 62, 818-825, 10.1111/myc.12955.
- Arielle Butts; Kristy Koselny; Yeissa Chabrier-Roselló; Camile P. Semighini; Jessica Cs Brown; Xuying Wang; Sivakumar Annadurai; Louis DiDone; Julie Tabroff; Wayne E. Childers; et al. Estrogen Receptor Antagonists Are Anti-Cryptococcal Agents That Directly Bind EF Hand Proteins and Synergize with Fluconazole In Vivo. mBio 2014, 5, e00765-13, 10.1128/mBio.00765-13.
- Juliana Santos-Gandelman; Marcio L. Rodrigues; Alice Machado-Silva; Future perspectives for cryptococcosis treatment. Expert Opinion on Therapeutic Patents 2018, 28, 625–634, 10.1080/13543776.2018.1503252.
- Kristy Koselny; Julianne Green; Lacey Favazzo; Virginia E. Glazier; Louis DiDone; Shea Ransford; Damian J. Krysan; Antitumor/Antifungal Celecoxib Derivative AR-12 is a Non-Nucleoside Inhibitor of the ANL-Family Adenylating Enzyme Acetyl CoA Synthetase. ACS Infectious Diseases 2016, 2, 268-280, 10.1021/acsinfecdis.5b00134.
- Kristy Koselny; Julianne Green; Louis DiDone; Justin P. Halterman; Annette W. Fothergill; Nathan P Wiederhold; Thomas F. Patterson; Melanie T. Cushion; Chad Rappelye; Melanie Wellington; et al. The celecoxib derivative AR-12 has broad spectrum antifungal activity in vitro and improves the activity of fluconazole in a murine model of cryptococcosis. Antimicrobial Agents and Chemotherapy 2016, 60, 7115–7127, 10.1128/aac.01061-16.
- Shyam Sundar; Jaya Chakravarty; An update on pharmacotherapy for leishmaniasis. Expert Opinion on Pharmacotherapy 2015, 16, 237-252, 10.1517/14656566.2015.973850.
- Ranga Rao Ravu; Ying-Lien Chen; Melissa R. Jacob; Xuewen Pan; Ameeta K. Agarwal; Shabana I. Khan; Joseph Heitman; Alice M. Clark; Xing-Cong Li; Synthesis and antifungal activities of miltefosine analogs.. Bioorganic & Medicinal Chemistry Letters 2013, 23, 4828-31, 10.1016/j.bmcl.2013.06.096.
- Fred Widmer; Lesley C. Wright; Daniel Obando; Rosemary Handke; Ranjini Ganendren; David H. Ellis; Tania C. Sorrell; Hexadecylphosphocholine (Miltefosine) Has Broad-Spectrum Fungicidal Activity and Is Efficacious in a Mouse Model of Cryptococcosis. Antimicrobial Agents and Chemotherapy 2006, 50, 414-421, 10.1128/aac.50.2.414-421.2006.
- Cristina De Castro Spadari; Taissa Vila; Sonia Rozental; Kelly Ishida; Miltefosine Has a Postantifungal Effect and Induces Apoptosis inCryptococcusYeasts. Antimicrobial Agents and Chemotherapy 2018, 62, 1–11, 10.1128/aac.00312-18.
- Cristina De Castro Spadari; Fernanda Walt Mendes Da Silva De Bastiani; Luciana Biagini Lopes; Kelly Ishida; Alginate nanoparticles as non-toxic delivery system for miltefosine in the treatment of candidiasis and cryptococcosis. International Journal of Nanomedicine 2019, 14, 5187-5199, 10.2147/IJN.S205350.
- Nathan P Wiederhold; Laura K. Najvar; Rosie Bocanegra; William R. Kirkpatrick; Tania C. Sorrell; Thomas F. Patterson; Limited Activity of Miltefosine in Murine Models of Cryptococcal Meningoencephalitis and Disseminated Cryptococcosis. Antimicrobial Agents and Chemotherapy 2012, 57, 745-750, 10.1128/AAC.01624-12.
- Anran Qian; Yazhou Zheng; Ruilian Wang; Jianhai Wei; YongMei Cui; Xufeng Cao; Yushe Yang; Design, synthesis, and structure-activity relationship studies of novel tetrazole antifungal agents with potent activity, broad antifungal spectrum and high selectivity. Bioorganic & Medicinal Chemistry Letters 2018, 28, 344-350, 10.1016/j.bmcl.2017.12.040.
- Sheng-Qiang Wang; Yan-Fang Wang; Zhi Xu; Tetrazole hybrids and their antifungal activities. European Journal of Medicinal Chemistry 2019, 170, 225-234, 10.1016/j.ejmech.2019.03.023.
- Nathan P Wiederhold; The antifungal arsenal: alternative drugs and future targets. International Journal of Antimicrobial Agents 2018, 51, 333-339, 10.1016/j.ijantimicag.2017.09.002.
- John R. Perfect; The antifungal pipeline: a reality check. Nature Reviews Drug Discovery 2017, 16, 603-616, 10.1038/nrd.2017.46.
- Shawn R. Lockhart; Annette W. Fothergill; Naureen Iqbal; Carol B. Bolden; Nina T. Grossman; Edward P. Garvey; Stephen R. Brand; William J. Hoekstra; Robert J. Schotzinger; Elizabeth Ottinger; et al. The Investigational Fungal Cyp51 Inhibitor VT-1129 Demonstrates Potent In Vitro Activity against Cryptococcus neoformans and Cryptococcus gattii. Antimicrobial Agents and Chemotherapy 2016, 60, 2528-2531, 10.1128/AAC.02770-15.
- Kirsten Nielsen; Priya Vedula; Kyle D. Smith; David B. Meya; Edward P. Garvey; William J. Hoekstra; Robert J. Schotzinger; David Boulware; Activity of VT-1129 against Cryptococcus neoformans clinical isolates with high fluconazole MICs. Medical Mycology 2017, 55, 453–456, 10.1093/mmy/myw089.
- Nathan P Wiederhold; Hoja P Patterson; Bich Hue Tran; Christopher M Yates; Robert J Schotzinger; Edward P Garvey; Fungal-specific Cyp51 inhibitor VT-1598 demonstrates in vitro activity against Candida and Cryptococcus species, endemic fungi, including Coccidioides species, Aspergillus species and Rhizopus arrhizus. Journal of Antimicrobial Chemotherapy 2018, 73, 404-408, 10.1093/jac/dkx410.
- E. P. Garvey; A D Sharp; P A Warn; C M Yates; R J Schotzinger; The novel fungal CYP51 inhibitor VT-1598 is efficacious alone and in combination with liposomal amphotericin B in a murine model of cryptococcal meningitis. Journal of Antimicrobial Chemotherapy 2018, 73, 2815-2822, 10.1093/jac/dky242.