The exact connection between Alzheimer’s disease (AD) and type 2 diabetes is still in debate. However, poorly controlled blood sugar may increase the risk of developing Alzheimer’s. This relationship is so strong that some have called Alzheimer’s “diabetes of the brain” or “type 3 diabetes (T3D)”. Given more recent studies continue to indicate evidence linking T3D with AD, this state-of-the-art aimed to demonstrate the relationship between T3D and AD based on the fact that both the processing of amyloid-β (Aβ) precursor protein toxicity and the clearance of Aβ are attributed to impaired insulin signaling, and that insulin resistance mediates the dysregulation of bioenergetics and progress to AD.
- Alzheimer’s disease
- hypometabolism
- type 2 diabetes
- type 3 diabetes
- insulin resistance
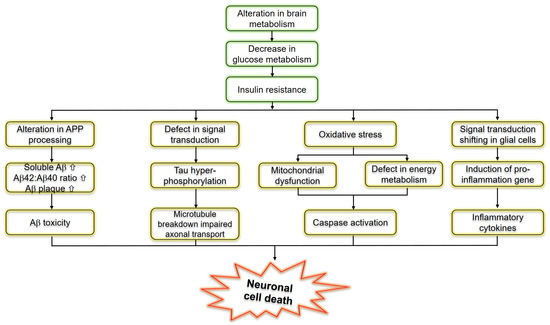
The Role of Type 3 Diabetes in Glucose Homeostasis
Type 3 Diabetes and Aβ Protein Pathology
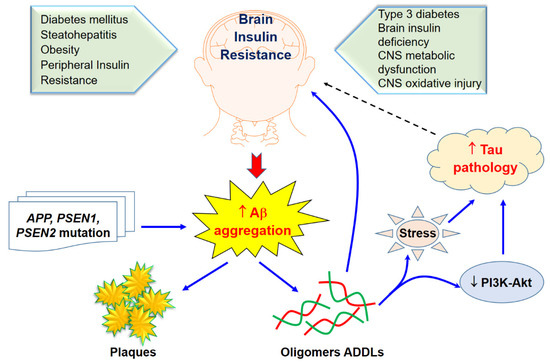
Type 3 Diabetes Regarding Alzheimer’s Disease
| Upstream Risk Factors | Metabolic Precursors | Pathways | Subclinical Pathology | Disease Outcome | |
|---|---|---|---|---|---|
| Social factors: stress, low socioeconomic status, certain ethnic and racial groups | Obesity Visceral Adiposity |
Vascular Processes Blood pressure and hypertension Hyperlipidemia Apolipoprotein E |
Cerebral blood flow Atherosclerosis |
Amyloid precursor proteins | Alzheimer’s disease |
| Poor diet: high in calories, fat and sugar, low in fiver | Inflammatory/Oxidative processes Inflammation Oxidative stress Endothelial function |
Neurofibrillary Tangles Amyloid B deposits |
|||
| Physical inactivity Genetics and family history |
Hyperglycemia Hyperinsulinemia |
Metabolic processes Insulin resistance Insulin-degrading enzyme Peroxisome proliferative-activated receptors |
|||
| Early childhood exposures in utero and birth weight | Brain and hippocampal atrophy White matter hyperintensities |
||||
Therapeutic Approaches to Type 3 Diabetes in Alzheimer’s Disease
| Compound | Potential Pathway | Study Design | Reference |
|---|---|---|---|
| DA5-CH | Reduces tau phosphorylation and normalizes theta rhythm | Injected intracerebroventricula (ICV), streptozotocin on rat | [89] |
| DA-JC1 | Antagonizing circadian rhythm disorders induced by Aβ31–35 | ICV, amyloid(31–35) AD model | [90] |
| DA5-CH | Improved of hippocampal synaptic plasticity and activation of the PI3K/AKT signaling pathway | APP/PS1 mouse model of AD | [91] |
| DA-CH3 | Reduced ER stress and apoptotic signaling, reduced amyloid plaque load in the brain | APP/PS1 mouse model of AD | [92] |
| Insulin | Prevention of Aβ oligomer induced synapse loss and insulin receptor reduction, amelioration of PKR-mediated ER stress | Rat hippocampal neuronal cultures | [93,94] |
| Insulin | AD patients that are not ε4 carriers have reduced sensitivity to insulin, effecting cognitive performance | AD patients homozygous or not for the ApoE-ε4 allele and normal subjects intravenously injected | [95] |
| Insulin | Improved verbal memory in MCI AD ε4-subjects after acute insulin administration, but not in ε4 carriers | AD patients homozygous or not for the ApoE-ε4 allele, MCI patients and most subjects intranasally administrated | [96,97] |
| Insulin | Chromic intranasal insulin doses enhanced selective attention, retention of new information and functional status of MCI and early AD subjects | AD patients, MCI patients and normal subjects intranasally administrated | [98] |
| Insulin | Only women presented improved working memory after treatment | Healthy men and woman intranasally administrated | [99] |
| Liraglutide | Reduction of tau phosphorylation; protection of insulin reception and synapse loss in a c-AMP dependent manner | Cynomolgus monkeys ICV with Aβ oligomer | [100] |
| Liraglutide | Improvement of memory deficits in novel object recognition test and fear conditioning | Swiss mice injected ICV with Aβ oligomer | [100] |
| Liraglutide | Restored memory deficits in object recognition test and Morris water maze; enhanced LTP; reduced microglial activation; diminished Aβ plaque load | APP/PSEN1 mice | [101,102] |
| Exendin-4 | Decrease in the inhibitory phosphorylation of Ser312IRS1, Ser66IRS1 of INK, while restoring activating Tyr465 IRS1 phosphorylation | Rat hippocampal neural cultures | [69] |
| Exendin-4 | Improvement of spatial memory in the Morris water maze; reduced amyloid plaque LOAD | APP/PS1 mice | [69] |
| Exedin4- Liraglutide | eIF2α phosphorylation reduction | Rat hippocampal neural cultures, APP/PS1 mice, cynomolgus monkeys injected ICV with Aβ oligomer | [94] |
| GLP-1 Exendin-4 | Reduction of neural excitotoxicity | Rat hippocampal neural cultures, rats injected on the basal nucleus with ibotenic acid | [103] |
| Rosiglitazone | Reversal of memory deficits in objects recognition test and the Morris water maze; Aβ levels reduction | AD transgenic mice J20 line | [104] |
- Herrup, K.; Yang, Y. Cell cycle regulation in the postmitotic neuron: Oxymoron or new biology? Nat. Rev. Neurosci. 2007, 8, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Gubellini, P.; Picconi, B.; di Filippo, M.; Calabresi, P. Downstream mechanisms triggered by mitochondrial dysfunction in the basal ganglia: From experimental models to neurodegenerative diseases. Biochim. Biophys. Acta 2010, 1802, 151–161. [Google Scholar] [CrossRef]
- Apelt, J.; Mehlhorn, G.; Schliebs, R. Insulin-sensitive GLUT4 glucose transporters are colocalized with GLUT3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. J. Neurosci. Res. 1999, 57, 693–705. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.-X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Holscher, C. Common pathological processes in Alzheimer disease and type 2 diabetes: A review. Brain Res. Rev. 2007, 56, 384–402. [Google Scholar] [CrossRef]
- Goldstein, B.J. Insulin resistance as the core defect in type 2 diabetes mellitus. Am. J. Cardiol. 2002, 90, 3g–10g. [Google Scholar] [CrossRef]
- Mielke, J.G.; Taghibiglou, C.; Liu, L.; Zhang, Y.; Jia, Z.; Adeli, K.; Wang, Y.T. A biochemical and functional characterization of diet-induced brain insulin resistance. J. Neurochem. 2005, 93, 1568–1578. [Google Scholar] [CrossRef]
- Hardigan, T.; Ward, R.; Ergul, A. Cerebrovascular complications of diabetes: Focus on cognitive dysfunction. Clin. Sci. (Lond.) 2016, 130, 1807–1822. [Google Scholar] [CrossRef]
- Hoyer, S. The brain insulin signal transduction system and sporadic (type II) Alzheimer disease: An update. J. Neural Transm. (Vienna, Austria: 1996) 2002, 109, 341–360. [Google Scholar] [CrossRef] [PubMed]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guénette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 35–66. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Martins, R.N.; Racchi, M.; Craft, S.; Helmerhorst, E. Amyloid beta antagonizes insulin promoted secretion of the amyloid beta protein precursor. J. Alzheimer’s Dis. Jad. 2002, 4, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.H.; Kar, S.; Quirion, R. Insulin-like growth factor-1-induced phosphorylation of the forkhead family transcription factor FKHRL1 is mediated by Akt kinase in PC12 cells. J. Biol. Chem. 2000, 275, 39152–39158. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Gouras, G.K.; Wang, R.; Gross, R.S.; Beal, M.F.; Greengard, P.; Xu, H. Stimulation of β-Amyloid Precursor Protein Trafficking by Insulin Reduces Intraneuronal β-Amyloid and Requires Mitogen-Activated Protein Kinase Signaling. J. Neurosci. 2001, 21, 2561–2570. [Google Scholar] [CrossRef] [PubMed]
- Delikkaya, B.; Moriel, N.; Tong, M.; Gallucci, G.; de la Monte, S.M. Altered expression of insulin-degrading enzyme and regulator of calcineurin in the rat intracerebral streptozotocin model and human apolipoprotein E-ε4–associated Alzheimer’s disease, Alzheimer’s & Dementia: Diagnosis. Assess. Dis. Monit. 2019, 11, 392–404. [Google Scholar]
- Mittal, K.; Mani, R.J.; Katare, D.P. Type 3 Diabetes: Cross Talk between Differentially Regulated Proteins of Type 2 Diabetes Mellitus and Alzheimer’s Disease. Sci. Rep. 2016, 6, 25589. [Google Scholar] [CrossRef]
- Kroner, Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern. Med. Rev. A J. Clin. Ther. 2009, 14, 373–379. [Google Scholar]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- de la Monte, S.M. Type 3 diabetes is sporadic Alzheimer’s disease: Mini-review. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2014, 24, 1954–1960. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kareholt, I.; Winblad, B.; Helkala, E.L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [CrossRef] [PubMed]
- Razay, G.; Vreugdenhil, A.; Wilcock, G. Obesity, abdominal obesity and Alzheimer disease. Dement. Geriatr. Cogn. Disord. 2006, 22, 173–176. [Google Scholar] [CrossRef]
- Kullmann, S.; Heni, M.; Hallschmid, M.; Fritsche, A.; Preissl, H.; Haring, H.U. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol. Rev. 2016, 96, 1169–1209. [Google Scholar] [CrossRef]
- Gabbouj, S.; Ryhänen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; et al. Altered Insulin Signaling in Alzheimer’s Disease Brain—Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629. [Google Scholar] [CrossRef]
- Lillioja, S.; Mott, D.M.; Spraul, M.; Ferraro, R.; Foley, J.E.; Ravussin, E.; Knowler, W.C.; Bennett, P.H.; Bogardus, C. Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. Prospective studies of Pima Indians. N. Engl. J. Med. 1993, 329, 1988–1992. [Google Scholar] [CrossRef]
- Li, J.; Bai, L.; Wei, F.; Zhao, J.; Wang, D.; Xiao, Y.; Yan, W.; Wei, J. Therapeutic Mechanisms of Herbal Medicines Against Insulin Resistance: A Review. Front. Pharmacol. 2019, 10, 661. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Biragyn, A.; Masharani, U.; Frassetto, L.; Petersen, R.C.; Miller, B.L.; Goetzl, E.J. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 589–596. [Google Scholar] [CrossRef]
- Fontaine, J.F.; Barbosa-Silva, A.; Schaefer, M.; Huska, M.R.; Muro, E.M.; Andrade-Navarro, M.A. MedlineRanker: Flexible ranking of biomedical literature. Nucleic Acids Res. 2009, 37, W141–W146. [Google Scholar] [CrossRef]
- Wang, G. Raison d’être of insulin resistance: The adjustable threshold hypothesis. J. R. Soc. Interface 2014, 11, 20140892. [Google Scholar] [CrossRef]
- Nisr, R.B.; Affourtit, C. Insulin acutely improves mitochondrial function of rat and human skeletal muscle by increasing coupling efficiency of oxidative phosphorylation. Biochim. Biophys. Acta 2014, 1837, 270–276. [Google Scholar] [CrossRef]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [PubMed]
- Bucht, G.; Adolfsson, R.; Lithner, F.; Winblad, B. Changes in blood glucose and insulin secretion in patients with senile dementia of Alzheimer type. Acta Med. Scand. 1983, 213, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Matioli, M.N.P.S.; Nitrini, R. Mechanisms linking brain insulin resistance to Alzheimer’s disease. Dement. Neuropsychol. 2015, 9, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 9078–9089. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, R.; Tran, A.; Ishimwe, E.; Denner, L.; Dave, N.; Oddo, S.; Dineley, K.T. Central insulin dysregulation and energy dyshomeostasis in two mouse models of Alzheimer’s disease. Neurobiol. Aging 2017, 58, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, H.H.; Chi, T.; Shin, A.C.; Lindtner, C.; Hsieh, W.; Ehrlich, M.; Gandy, S.; Buettner, C. Increased susceptibility to metabolic dysregulation in a mouse model of Alzheimer’s disease is associated with impaired hypothalamic insulin signaling and elevated BCAA levels. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 851–861. [Google Scholar] [CrossRef]
- Long-Smith, C.M.; Manning, S.; McClean, P.L.; Coakley, M.F.; O’Halloran, D.J.; Holscher, C.; O’Neill, C. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-beta plaque and glial pathology in a mouse model of Alzheimer’s disease. Neuromol. Med. 2013, 15, 102–114. [Google Scholar] [CrossRef]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Abeta oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef]
- Clarke, J.R.; Lyra, E.S.N.M.; Figueiredo, C.P.; Frozza, R.L.; Ledo, J.H.; Beckman, D.; Katashima, C.K.; Razolli, D.; Carvalho, B.M.; Frazao, R.; et al. Alzheimer-associated Abeta oligomers impact the central nervous system to induce peripheral metabolic deregulation. Embo Mol. Med. 2015, 7, 190–210. [Google Scholar] [CrossRef]
- Tanokashira, D.; Fukuokaya, W.; Taguchi, A. Involvement of insulin receptor substrates in cognitive impairment and Alzheimer’s disease. Neural Regen. Res. 2019, 14, 1330–1334. [Google Scholar] [PubMed]
- Freude, S.; Hettich, M.M.; Schumann, C.; Stohr, O.; Koch, L.; Kohler, C.; Udelhoven, M.; Leeser, U.; Muller, M.; Kubota, N.; et al. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 3315–3324. [Google Scholar]
- Taguchi, A.; Wartschow, L.M.; White, M.F. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science (N. Y.) 2007, 317, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Killick, R.; Scales, G.; Leroy, K.; Causevic, M.; Hooper, C.; Irvine, E.E.; Choudhury, A.I.; Drinkwater, L.; Kerr, F.; Al-Qassab, H.; et al. Deletion of Irs2 reduces amyloid deposition and rescues behavioural deficits in APP transgenic mice. Biochem. Biophys. Res. Commun. 2009, 386, 257–262. [Google Scholar] [CrossRef]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a Type 3 Diabetes? A critical appraisal. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Pham, Q.T.; Luong, T.N.H.; Le, H.K.; Vo, V.G. Potential Antidiabetic Activity of Extracts and Isolated Compound from Adenosma bracteosum (Bonati). Biomolecules 2020, 10, 201. [Google Scholar] [CrossRef]
- Giau, V.V.; Wu, S.Y.; Jamerlan, A.; An, S.S.A.; Kim, S.; Hulme, J. Gut Microbiota and Their Neuroinflammatory Implications in Alzheimer’s Disease. Nutrients 2018, 10, 1765. [Google Scholar] [CrossRef]
- de Matos, A.M.; de Macedo, M.P.; Rauter, A.P. Bridging Type 2 Diabetes and Alzheimer’s Disease: Assembling the Puzzle Pieces in the Quest for the Molecules With Therapeutic and Preventive Potential. Med. Res. Rev. 2018, 38, 261–324. [Google Scholar] [CrossRef]
- Bagyinszky, E.; Giau, V.V.; Shim, K.; Suk, K.; An, S.S.A.; Kim, S. Role of inflammatory molecules in the Alzheimer’s disease progression and diagnosis. J. Neurol. Sci. 2017, 376, 242–254. [Google Scholar] [CrossRef]
- van Giau, V.; An, S.S.A.; Hulme, J.P. Mitochondrial therapeutic interventions in Alzheimer’s disease. J. Neurol. Sci. 2018, 395, 62–70. [Google Scholar] [CrossRef]
- Ayaz, M.; Sadiq, A.; Junaid, M.; Ullah, F.; Ovais, M.; Ullah, I.; Ahmed, J.; Shahid, M. Flavonoids as Prospective Neuroprotectants and Their Therapeutic Propensity in Aging Associated Neurological Disorders. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Canhada, S.; Castro, K.; Perry, I.S.; Luft, V.C. Omega-3 fatty acids’ supplementation in Alzheimer’s disease: A systematic review. Nutr. Neurosci. 2018, 21, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Ajith, T.A. A Recent Update on the Effects of Omega-3 Fatty Acids in Alzheimer’s Disease. Curr. Clin. Pharmacol. 2018, 13, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Broom, G.M.; Shaw, I.C.; Rucklidge, J.J. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition (Burbank, Los Angeles County, Calif.) 2019, 60, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Bagyinszky, E.; An, S.S.A.; Kim, S.Y. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 1723–1737. [Google Scholar] [CrossRef]
- Stoykovich, S.; Gibas, K. APOE ε4, the door to insulin-resistant dyslipidemia and brain fog? A case study. Alzheimers Dement. (Amst) 2019, 11, 264–269. [Google Scholar] [CrossRef]
- Zhao, N.; Liu, C.C.; van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129.e5. [Google Scholar] [CrossRef]
- Frederiksen, K.S.; Gjerum, L.; Waldemar, G.; Hasselbalch, S.G. Effects of Physical Exercise on Alzheimer’s Disease Biomarkers: A Systematic Review of Intervention Studies. J. Alzheimer’s Dis. 2018, 61, 359–372. [Google Scholar] [CrossRef]
- Li, C.; Liu, W.; Li, X.; Zhang, Z.; Qi, H.; Liu, S.; Yan, N.; Xing, Y.; Holscher, C.; Wang, Z. The novel GLP-1/GIP analogue DA5-CH reduces tau phosphorylation and normalizes theta rhythm in the icv. STZ rat model of AD. Brain Behav. 2020, 10, e01505. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, R.; Hou, X.; Wang, C.; Guo, S.; Ning, N.; Sun, C.; Yuan, Y.; Li, L.; Hölscher, C.; et al. DA-JC1 improves learning and memory by antagonizing Aβ31–35-induced circadian rhythm disorder. Mol. Brain 2019, 12, 14. [Google Scholar] [CrossRef]
- Cao, Y.; Holscher, C.; Hu, M.M.; Wang, T.; Zhao, F.; Bai, Y.; Zhang, J.; Wu, M.N.; Qi, J.S. DA5-CH, a novel GLP-1/GIP dual agonist, effectively ameliorates the cognitive impairments and pathology in the APP/PS1 mouse model of Alzheimer’s disease. Eur. J. Pharmacol. 2018, 827, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Panagaki, T.; Gengler, S.; Holscher, C. The Novel DA-CH3 Dual Incretin Restores Endoplasmic Reticulum Stress and Autophagy Impairments to Attenuate Alzheimer-Like Pathology and Cognitive Decrements in the APPSWE/PS1DeltaE9 Mouse Model. J. Alzheimer’s Dis. 2018, 66, 195–218. [Google Scholar] [CrossRef] [PubMed]
- de Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Asthana, S.; Cook, D.G.; Baker, L.D.; Cherrier, M.; Purganan, K.; Wait, C.; Petrova, A.; Latendresse, S.; Watson, G.S.; et al. Insulin dose-response effects on memory and plasma amyloid precursor protein in Alzheimer’s disease: Interactions with apolipoprotein E genotype. Psychoneuroendocrinology 2003, 28, 809–822. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Cherrier, M.M.; Schellenberg, G.D.; Frey, W.H., 2nd; et al. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimer’s Dis. 2008, 13, 323–331. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Frey, W.H., 2nd; Baker, L.D.; Cholerton, B.; Keeling, M.L.; Belongia, D.A.; Fishel, M.A.; Plymate, S.R.; Schellenberg, G.D.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef]
- Benedict, C.; Kern, W.; Schultes, B.; Born, J.; Hallschmid, M. Differential sensitivity of men and women to anorexigenic and memory-improving effects of intranasal insulin. J. Clin. Endocrinol. Metab. 2008, 93, 1339–1344. [Google Scholar] [CrossRef]
- Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; Lyra, E.S.N.M.; Brito-Moreira, J.; Boehnke, S.E.; Winterborn, A.; Coe, B.C.; Lablans, A.; Vital, J.F.; et al. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol. 2018, 245, 85–100. [Google Scholar] [CrossRef]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Holscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Holscher, C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology 2014, 76 Pt A, 57–67. [Google Scholar] [CrossRef]
- Perry, T.; Haughey, N.J.; Mattson, M.P.; Egan, J.M.; Greig, N.H. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J. Pharmacol. Exp. Ther. 2002, 302, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Escribano, L.; Simon, A.M.; Gimeno, E.; Cuadrado-Tejedor, M.; de Maturana, R.L.; Garcia-Osta, A.; Ricobaraza, A.; Perez-Mediavilla, A.; del Rio, J.; Frechilla, D. Rosiglitazone rescues memory impairment in Alzheimer’s transgenic mice: Mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2010, 35, 1593–1604. [Google Scholar] [CrossRef]
This entry is adapted from the peer-reviewed paper 10.3390/ijms21093165