β-adrenergic receptors (β-ARs) play a major role in the physiological regulation of cardiac function through signaling routes tightly controlled by G protein-coupled receptor kinases (GRKs).
- β-adrenergic receptors
- Epac1
- cAMP
- G protein-coupled receptor kinases
- signaling
1. Introduction
G-protein-coupled receptors (GPCR) represent one of the largest superfamilies of transmembrane receptors and are involved in a wide variety of biological functions. Among the numerous GPCRs expressed in the cardiovascular system, β-adrenergic receptors (β-ARs) represent the most powerful system that regulates cardiac function and acutely increases the output of the heart[1]. In the classical paradigm, acute stimulation of cardiac β-AR by catecholamines (adrenaline, noradrenaline) induces the intracellular production of cyclic AMP (cAMP) and protein kinase A (PKA) activation, which plays a key role in the regulation of the contraction and relaxation of cardiac myocytes[2]. In addition to heterodimeric G protein activation and their downstream effectors, GPCRs promote receptor phosphorylation by G protein-coupled receptor kinases (GRKs) in an agonist dose-dependent fashion[3][4]. This process induces receptor desensitization through the binding of the scaffold protein β-arrestin (β-arr) to terminate receptor signaling.
Despite their capacity to confer sensitivity to acute adrenergic stimuli involved in physiological signaling, β-ARs also form a crucial part of the stress response pathways linked to and involved in cardiovascular disease, including heart failure (HF), a leading cause of death worldwide. Indeed, the excessive sympathetic nervous system activity observed in HF can promote the continued stimulation of cardiac β-ARs, which triggers adverse effects including over proportional increases in energy consumption, cell death, fibrosis, cardiomyocyte hypertrophy, and arrhythmia [5]. β-AR blockers are a mainstay in the therapy of HF, but the morbidity and mortality associated with HF still continue to rise. Therefore, it is important to elucidate the molecular players that couple the β-AR signaling route to pathological cardiac remodeling leading to HF, since novel findings might have therapeutic implications for novel treatment.
In addition to canonical receptor desensitization and downregulation, GRK2 and GRK5 are critical mediators of the molecular alterations that contribute to HF. The exchange protein directly activated by cAMP 1 (Epac1) is coupled to β-Ars, and various studies have revealed the involvement of this cAMP-binding protein in cardiac remodeling and HF [6][7][8][9][10].
2. Epac1 and GRK Molecular Complex Formation in Cardiac Remodeling
As previously mentioned, chronic β-adrenergic activation results in desensitization and internalization of β-AR, which is accompanied by a decreased fight-or-flight response and detrimental changes, such as cardiomyocyte hypertrophy, apoptosis, and inflammation. Both GRKs and PKA induce the phosphorylation of agonist-activated β-AR in order to terminate signaling. This drives the recruitment of β-arr to the β-AR receptor, which sterically prevents further G-protein coupling to the receptor while promoting β-AR internalization[11][12]. Besides the desensitization and internalization of activated receptors, β-arr also acts as a nodal point, and further initiates the activation of many intracellular signaling routes independently of G protein activity through their function as scaffold proteins[13]. In this regard, β-arr in the heart may undergo GRK-arrestin signaling and play an important role in regulating normal and compromised cardiomyocyte function. Here below, we will describe how macromolecular complexes composed of β-arr, Epac1 and other signaling molecules regulate prohypertrophic signaling, and may be involved in the pathogenesis of HF.
2.1. Epac1, CaMKII and β-Arrestin Complex
The formation of a β-arr–Epac1 complex was initially reported in the heart[14]. Biochemical studies demonstrated that Epac1 constitutively interacts through its RA domain with the scaffold protein β-arrestin2 (β-arr2) in the cytosol of cardiomyocytes during basal conditions [14][15]. Although both β1-AR and β2-AR activate Epac1, only β1-AR stimulation allows the recruitment of Epac1-β-arr2 molecular complex to the plasma membrane, a process involving the activity of microtubules[16]. It is suggested that GRK-induced β1-AR phosphorylation resulting in β-arr recruitment might also stabilize Epac1 at the plasma membrane [14][17]. Consistent with studies showing that β-arr can facilitate binding to distinct signaling partners[12][18], the interaction of β-arr with the C-terminal tail of the β1-AR but not β2-AR induces a conformational change in β-arr that favors its interaction with Epac1[14]. In close proximity to the β1-AR complex, Epac1 promotes the activation of the Ca2+ sensitive protein CaMKII, which promotes the phosphorylation of the histone deacetylase 4 (HDAC4). Phosphorylated HDAC4 is extruded out of the nucleus and relieves HDAC4 inhibition on the hypertrophic transcription factor, myocyte enhancer factor 2 (MEF2)[19][15].
The involvement of PKA in CaMKII activation seems to depend on the length and intensity of the stimulus. Indeed, a recent study using FRET-based biosensor showed that inhibition of PKA prevented acute stimulation of β1-AR-induced CaMKII activation[20]. However, CaMKII activity induced by prolonged activation of β-1AR was still maintained in the presence of PKA inhibitors[21]. Based on these observations, one could speculate that during chronic activation of cardiac β1-AR, there is a switch of β1-AR signaling from physiological cAMP-PKA activity to Epac1-CaMKII activity that promotes the development of cardiac remodeling and HF (Figure 1). Accordingly, expression and activity of CaMKII are increased during cardiac hypertrophy and HF[22][23]. In addition, chronic inhibition or gene deletion of CaMKII appears to have little effect on basal cardiac function or on acute responses to β-adrenergic stimulation but confer protection against pathological stresses known to be associated with chronic sympathetic activation[24].
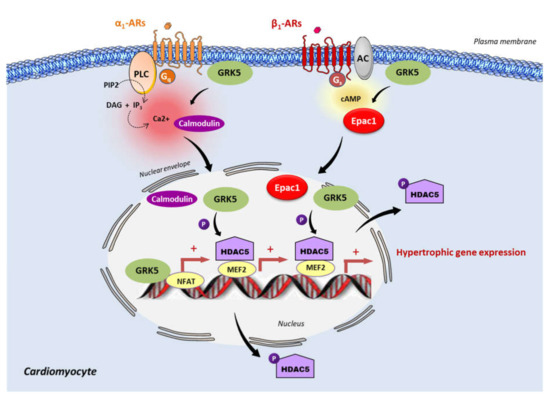
Figure 1. Epac1 and GRK5 non-canonical hypertrophic signaling. Activation of the Gαq coupled receptor, α1-adrenergic receptor (α1-AR), promotes the intracellular elevation of Ca2+ through the phospholipase C (PLC) and the subsequent activation of calmodulin. GRK5 associates with Ca2+-calmodulin, leading to the translocation of GRK5 to the nucleus, wherein it promotes HDAC5 phosphorylation and subsequent myocyte enhancer factor 2 (MEF2) activation. During the chronic stimulation of β1-adrenergic receptor (β1-AR), Epac1 interacts with GRK5 and the Epac1-GRK5 molecular complex is exported to the nucleus of cardiomyocytes. There, GRK5 phosphorylates HDAC5, leading to its nuclear export, and thereby derepressing MEF2 transcriptional activity. GRK5 also acts as a coactivator of the nuclear factor of activated T cells (NFAT). MEF2 and NFAT are crucial transcription factors that promote cardiac hypertrophy.
Of particular importance, the interaction of Epac1 with β-arr2 is also regulated by the presence of the cAMP-specific phosphodiesterase 4 (PDE4) variant, PDE4D5. Indeed, Epac1 and PDE4D5 compete by steric hindrance for binding to β-arr2. Blocking the formation of the PDE4D5–β-arr molecular complex allows the translocation of the Epac1–β-arr complex to the activated β2-AR. Consequently, the β2-AR signaling switches to a β1-AR-like pro-hypertrophic signaling, and increases cardiomyocyte remodeling[15]. These data indicate that the differential interaction of Epac1 with β-arr2 contributes to the specificity of β1-AR and β2-AR signaling, and Epac1 compartmentalization participates in the distinct functions of the β-AR subtypes[17][15]. Interestingly, the interaction of β-arr seems not to be limited to Epac1, since a more recent study reported that the Epac2 isoform can directly interact with β-arr1 to regulate insulin secretion in pancreatic β cells[25].
As expected, Epac1 can undergo post-translational modifications, such as phosphorylation, that can regulate its activity. In this line, it has been demonstrated that in dorsal root ganglion neurons GRK2 controls Epac1-to-Rap1 signaling by phosphorylating Epac1 at Ser-108 in its DEP domain. This mechanism prevents the translocation of Epac1 to the plasma membrane in response to cAMP elevation, and underlies the protective effect of GRK2 on chronic inflammatory pain[26]. Whether such a non-canonical effect of GRK2 on Epac1 activity occurs in the context of cardiac remodeling has yet to be investigated.
2.2. Epac1 and GRK5
Since Epac1 and GRK5 are linked to hypertrophic signaling, recent studies have investigated their possible interaction in the regulation of this process. Recently, Laudette and collaborators (2019) showed that cardiac remodeling induced by chronic injection of the synthetic β-AR agonist, isoprenaline (ISO), increased GRK2 and GRK5 expression protein levels in mouse hearts[6]. Interestingly, the upregulation of GRK5, but not GRK2, was decreased in animals treated with AM-001, a specific pharmacological inhibitor of Epac1, suggesting that Epac1 specifically targets GRK5[6]. At the molecular level, ISO increased Epac1–GRK5 interaction and promoted GRK5 nuclear import, while Epac1 inhibition with AM-001 prevented GRK5 nuclear translocation, to induce the nuclear accumulation of HDAC5. GRK5 acted as a downstream effector of Epac1 since the knock-down of GRK5 blocked the stimulating effect of Epac1 on prohypertrophic signaling. Following β-AR stimulation, Epac1 activation induces GRK5 nuclear import and HDAC5 nuclear export to promote prohypertrophic gene expression. Therefore, Epac1 seems to be required for non-canonical nuclear roles of GRK5 in maladaptive cardiac remodeling (Figure 1). Interestingly, it has been reported that GRK4 promotes cardiomyocyte apoptosis through the phosphorylation of HDAC4 during myocardial infarction[27]. Whether Epac1 functions with GRK4 to regulate cardiomyocyte death has yet to be investigated[27].
Adding complexity to the matter, a recent finding investigated the role of the synapse-associated protein 97 (SAP97) in β-AR signaling[20]. SAP97 is a multifunctional scaffold protein that binds to the C-terminal PDZ motif of β1-AR [28][29]. Xu and colleagues (2020) reported that the β1-AR-SAP97 molecular complex was reduced in HF. In addition, the authors demonstrated that cardiac-specific deletion of SAP97 yielded to spontaneous development of cardiomyopathy and exacerbated cardiac dysfunction induced by chronic β-AR stimulation and myocardial pressure overload in mice[20]. Mechanistic studies showed that loss of SAP97 as observed in HF promoted the recruitment of β-arr2 and CaMKII to β1-AR and switched on the β1-AR signaling to Epac-dependent CaMKII activity[20]. Yet, it was shown that GRK5 and not GRK2 enhanced ISO-induced dissociation of SAP97 from β1-AR, thereby facilitating the activation of the Epac–CaMKII axis and its detrimental functional and structural remodeling (Figure 2). At present, it is still unknown which Epac isoform is involved in this process but given the ascertained role of Epac1 in cardiac hypertrophy, one could imagine that the Epac1 isoform could be involved in this specific signaling.
Another direction worth pursuing would be to examine the precise molecular mechanisms by which Epac mediates the activation of the Ca2+ sensitive protein, CaMKII, upon β1-AR stimulation. Does it depend on the activity of the downstream effector of Epac, the small GTPase Rap? Interestingly, a few years after the discovery of Epac, a study performed in HEK293 and neuroblastoma cells demonstrated that Epac activated the PLCε specifically through the Rap2 GTPase, resulting in the generation of inositol-1,4,5-trisphosphate (IP3) and the subsequent release of Ca2+ from intracellular stores[30]. Further studies showed that the Epac1–Rap axis could activate PLC, causing a Ca2+ increase via the IP3 receptor (IP3-R), to promote the activation of the Ca2+-sensitive dependent transcription factors involved in cardiac remodeling[19][31][32][33]. Alternatively, other downstream effectors of Epac1, including Rit and c-Jun NH2-terminal kinase (JNK), are activated by Rap1- and Rap2-independent mechanisms[34][35]. Whether these effectors are involved in the detrimental effect of Epac1 in cardiac remodeling has yet to be determined.
3. Conclusions
Evidence collected over the last decade indicates that GRK2, GRK5 and Epac1 contribute to the development and progression of HF, as illustrated here by experimental studies on pathological cardiac remodeling. A significant component of β-AR signaling is mediated through GRK and Epac1, and this pathway is particularly prominent under pathophysiological conditions. These promising results in terms of therapeutic innovation have stimulated the search for the identification of small molecules or peptides able to selectively inhibit the activity of a given GRK or Epac1 protein. In this line, several strategies have provided a proof of concept of the beneficial effect of blocking GRK2, GRK5 or Epac1 in animal models of HF. It is suggested that by normalizing β-AR signaling in human HF, GRK2-targeted therapeutic inhibition would restore the myocardial adrenergic reserve and improve cardiac function. Blocking the non-canonical action of GRK5 in the nucleus of cardiomyocytes prevents pathological hypertrophy. Finally, the pharmacological inhibition of Epac1 also ameliorates cardiac contractility, and attenuates cardiac remodeling and arrhythmia episodes by normalizing Ca2+ cycling. Whether GRK2, GRK5 or Epac1 inhibition would have a synergic effect on cardiac remodeling and HF has yet to be investigated. Therefore, further molecular studies are needed to characterize the GRK–Epac interactome and signalosome in subcellular compartments, so as to better understand how these proteins cross-talk to promote signaling alteration in HF.
This entry is adapted from the peer-reviewed paper 10.3390/cells10010154
References
- Rockman, H.A.; Koch, W.J.; Lefkowitz, R.J. Seven-transmembrane-spanning receptors and heart function. Nature 2002, 415, 206–212.
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205.
- Murga, C.; Arcones, A.C.; Cruces-Sande, M.; Briones, A.M.; Salaices, M.; Mayor, F. G Protein-Coupled Receptor Kinase 2 (GRK2) as a Potential Therapeutic Target in Cardiovascular and Metabolic Diseases. Front. Pharmacol. 2019, 10, 112.
- Pfleger, J.; Gresham, K.; Koch, W.J. G protein-coupled receptor kinases as therapeutic targets in the heart. Nat. Rev. Cardiol. 2019, 16, 612–622.
- El-Armouche, A.; Eschenhagen, T. Beta-adrenergic stimulation and myocardial function in the failing heart. Heart Fail. Rev. 2009, 14, 225–241.
- Laudette, M.; Coluccia, A.; Sainte-Marie, Y.; Solari, A.; Fazal, L.; Sicard, P.; Silvestri, R.; Mialet-Perez, J.; Pons, S.; Ghaleh, B.; et al. Identification of a pharmacological inhibitor of Epac1 that protects the heart against acute and chronic models of cardiac stress. Cardiovasc. Res. 2019, 115, 1766–1777.
- Lezoualc’h, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP Sensor EPAC Proteins and Their Role in Cardiovascular Function and Disease. Circ. Res. 2016, 118, 881–897.
- Laurent, A.-C.; Bisserier, M.; Lucas, A.; Tortosa, F.; Roumieux, M.; de Régibus, A.; Swiader, A.; Sainte-Marie, Y.; Heymes, C.; Vindis, C.; et al. Exchange protein directly activated by cAMP 1 promotes autophagy during cardiomyocyte hypertrophy. Cardiovasc. Res. 2015, 105, 55–64.
- Okumura, S.; Fujita, T.; Cai, W.; Jin, M.; Namekata, I.; Mototani, Y.; Jin, H.; Ohnuki, Y.; Tsuneoka, Y.; Kurotani, R.; et al. Epac1-dependent phospholamban phosphorylation mediates the cardiac response to stresses. J. Clin. Investig 2014, 124, 2785–2801.
- Métrich, M.; Lucas, A.; Gastineau, M.; Samuel, J.-L.; Heymes, C.; Morel, E.; Lezoualc’h, F. Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ. Res. 2008, 102, 959–965.
- De Lucia, C.; Eguchi, A.; Koch, W.J. New Insights in Cardiac β-Adrenergic Signaling During Heart Failure and Aging. Front. Pharmacol. 2018, 9, 904.
- Shukla, A.K.; Westfield, G.H.; Xiao, K.; Reis, R.I.; Huang, L.-Y.; Tripathi-Shukla, P.; Qian, J.; Li, S.; Blanc, A.; Oleskie, A.N.; et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 2014, 512, 218–222.
- Wang, J.; Gareri, C.; Rockman, H.A. G-Protein-Coupled Receptors in Heart Disease. Circ. Res. 2018, 123, 716–735.
- Mangmool, S.; Shukla, A.K.; Rockman, H.A. beta-Arrestin-dependent activation of Ca(2+)/calmodulin kinase II after beta(1)-adrenergic receptor stimulation. J. Cell Biol. 2010, 189, 573–587.
- Berthouze-Duquesnes, M.; Lucas, A.; Saulière, A.; Sin, Y.Y.; Laurent, A.-C.; Galés, C.; Baillie, G.; Lezoualc’h, F. Specific interactions between Epac1, β-arrestin2 and PDE4D5 regulate β-adrenergic receptor subtype differential effects on cardiac hypertrophic signaling. Cell. Signal. 2013, 25, 970–980.
- Dybkova, N.; Wagner, S.; Backs, J.; Hund, T.J.; Mohler, P.J.; Sowa, T.; Nikolaev, V.O.; Maier, L.S. Tubulin polymerization disrupts cardiac β-adrenergic regulation of late INa. Cardiovasc. Res. 2014, 103, 168–177.
- Boccella, N.; Paolillo, R.; Perrino, C. Epac1 inhibition as a novel cardioprotective strategy: Lights and shadows on GRK5 canonical and non-canonical functions. Cardiovasc. Res. 2019, 115, 1684–1686.
- Nuber, S.; Zabel, U.; Lorenz, K.; Nuber, A.; Milligan, G.; Tobin, A.B.; Lohse, M.J.; Hoffmann, C. β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 2016, 531, 661–664.
- Métrich, M.; Laurent, A.-C.; Breckler, M.; Duquesnes, N.; Hmitou, I.; Courillau, D.; Blondeau, J.-P.; Crozatier, B.; Lezoualc’h, F.; Morel, E. Epac activation induces histone deacetylase nuclear export via a Ras-dependent signalling pathway. Cell. Signal. 2010, 22, 1459–1468.
- Xu, B.; Li, M.; Wang, Y.; Zhao, M.; Morotti, S.; Shi, Q.; Wang, Q.; Barbagallo, F.; Teoh, J.-P.; Reddy, G.R.; et al. GRK5 Controls SAP97-Dependent Cardiotoxic β1 Adrenergic Receptor-CaMKII Signaling in Heart Failure. Circ. Res. 2020, 127, 796–810.
- Zhu, W.-Z.; Wang, S.-Q.; Chakir, K.; Yang, D.; Zhang, T.; Brown, J.H.; Devic, E.; Kobilka, B.K.; Cheng, H.; Xiao, R.-P. Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J. Clin. Investig. 2003, 111, 617–625.
- Dewenter, M.; Neef, S.; Vettel, C.; Lämmle, S.; Beushausen, C.; Zelarayan, L.C.; Katz, S.; von der Lieth, A.; Meyer-Roxlau, S.; Weber, S.; et al. Calcium/Calmodulin-Dependent Protein Kinase II Activity Persists During Chronic β-Adrenoceptor Blockade in Experimental and Human Heart Failure. Circ. Heart Fail. 2017, 10, e003840.
- Rokita, A.G.; Anderson, M.E. New therapeutic targets in cardiology: Arrhythmias and Ca2+/calmodulin-dependent kinase II (CaMKII). Circulation 2012, 126, 2125–2139.
- Anderson, M.E.; Brown, J.H.; Bers, D.M. CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 468–473.
- Barella, L.F.; Rossi, M.; Zhu, L.; Cui, Y.; Mei, F.C.; Cheng, X.; Chen, W.; Gurevich, V.V.; Wess, J. β-Cell-intrinsic β-arrestin 1 signaling enhances sulfonylurea-induced insulin secretion. J. Clin. Investig. 2019, 129, 3732–3737.
- Singhmar, P.; Huo, X.; Eijkelkamp, N.; Berciano, S.R.; Baameur, F.; Mei, F.C.; Zhu, Y.; Cheng, X.; Hawke, D.; Mayor, F.; et al. Critical role for Epac1 in inflammatory pain controlled by GRK2-mediated phosphorylation of Epac1. Proc. Natl. Acad. Sci. USA 2016, 113, 3036–3041.
- Li, L.; Fu, W.; Gong, X.; Chen, Z.; Tang, L.; Yang, D.; Liao, Q.; Xia, X.; Wu, H.; Liu, C.; et al. The role of G protein-coupled receptor kinase 4 in cardiomyocyte injury after myocardial infarction. Eur. Heart J. 2020.
- Nooh, M.M.; Chumpia, M.M.; Hamilton, T.B.; Bahouth, S.W. Sorting of β1-adrenergic receptors is mediated by pathways that are either dependent on or independent of type I PDZ, protein kinase A (PKA), and SAP97. J. Biol. Chem. 2014, 289, 2277–2294.
- Fu, Q.; Kim, S.; Soto, D.; de Arcangelis, V.; DiPilato, L.; Liu, S.; Xu, B.; Shi, Q.; Zhang, J.; Xiang, Y.K. A long lasting β1 adrenergic receptor stimulation of cAMP/protein kinase A (PKA) signal in cardiac myocytes. J. Biol. Chem. 2014, 289, 14771–14781.
- Schmidt, M.; Evellin, S.; Weernink, P.A.; von Dorp, F.; Rehmann, H.; Lomasney, J.W.; Jakobs, K.H. A new phospholipase-C-calcium signalling pathway mediated by cyclic AMP and a Rap GTPase. Nat. Cell Biol. 2001, 3, 1020–1024.
- Nash, C.A.; Brown, L.M.; Malik, S.; Cheng, X.; Smrcka, A.V. Compartmentalized cyclic nucleotides have opposing effects on regulation of hypertrophic phospholipase Cε signaling in cardiac myocytes. J. Mol. Cell. Cardiol. 2018, 121, 51–59.
- Pereira, L.; Ruiz-Hurtado, G.; Morel, E.; Laurent, A.-C.; Métrich, M.; Domínguez-Rodríguez, A.; Lauton-Santos, S.; Lucas, A.; Benitah, J.-P.; Bers, D.M.; et al. Epac enhances excitation-transcription coupling in cardiac myocytes. J. Mol. Cell. Cardiol. 2012, 52, 283–291.
- Zhang, L.; Malik, S.; Pang, J.; Wang, H.; Park, K.M.; Yule, D.I.; Blaxall, B.C.; Smrcka, A.V. Phospholipase Cε hydrolyzes perinuclear phosphatidylinositol 4-phosphate to regulate cardiac hypertrophy. Cell 2013, 153, 216–227.
- Hochbaum, D.; Tanos, T.; Ribeiro-Neto, F.; Altschuler, D.; Coso, O.A. Activation of JNK by Epac is independent of its activity as a Rap guanine nucleotide exchanger. J. Biol. Chem. 2003, 278, 33738–33746.
- Shi, G.-X.; Rehmann, H.; Andres, D.A. A novel cyclic AMP-dependent Epac-Rit signaling pathway contributes to PACAP38-mediated neuronal differentiation. Mol. Cell. Biol. 2006, 26, 9136–9147.