Exosomes are small bilipid layer enclosed extracellular vesicles, which were once considered as a cellular waste and functionless. These nano-vesicles of 30–150 nm in diameter carry specific proteins, lipids, functional mRNAs, and high amounts of non-coding RNAs (miRNAs, lncRNAs, and circRNAs). As the exosomes content is known to vary as per their originating and recipient cells, these vesicles can be utilized as a diagnostic biomarker for early disease detection.
- exosomes
- neurodegenerative diseases
- biomarkers
- Alzheimer’s disease
1. Exosomes: Biogenesis, Composition, and Their Diverse Functions
There exists a well-established process through which a variety of cells release different hormones or neurotransmitters, likewise, most cells have an evolutionarily conserved mechanism to secrete a myriad of membranous vesicles that are known as extracellular vesicles (EVs)[1]. These EVs are thought to be involved in clearing cellular junk, however, in recent years, the focus has been shifted to look into their auxiliary functions [2][3]. EVs are also involved in cell-to-cell communications by an exchange of different biomolecules including nucleic acids, lipids, and proteins, and thus held responsible for maintaining cellular homeostasis and in most cases results in the progression of current pathological manifestations[4]. Generally, EVs are highly heterogeneous in nature due to the associated cargo which is dependent on their releasing cell type. There are subtypes of extracellular vesicles based on their biogenesis and size, exosomes, and microvesicles[1][5]. Though there are some overlapping characteristics of both of them, the primary differences between exosomes and microvesicles are shown in Table 1.
Table 1. Key difference between exosomes and microvesicles.
| Exosomes | Microvesicles | |
|---|---|---|
| Size | 30–150 nm | 50–1000 nm |
| Morphology | Cup-shaped | Heterogeneous |
| Density | 1.1–1.2 g/mL | 1.08–1.19 g/mL |
| Origin | Multivesicular Endosomes (MVEs) | Plasma Membrane |
| Contents | Protein, miRNA, mRNA | Protein, miRNA, mRNA |
| Protein markers | Alix, Tsg101, CD 81, CD 82, CD63, CD 37, CD9 | Selectins, Integrins, CD40 |
Exosomes were first described in 1981[6] and were initially investigated by Johnstone et al.[7]. They noted the secretion of small vesicles (30–150 nm in diameter) by reticulocytes in sheep, explained their endocytic origin, and coined the term “exosomes” for these extracellular vesicles. The biogenesis of exosomes has been studied with several biochemical approaches in addition to transmission and immuno-electron microscopic (TEM/IEM) methods [8][9][10] (Figure 2). Briefly, multivesicular endosomes (MVEs) contain intraluminal vesicles (ILVs), which are formed by inward budding of the endosomal system. These ILVs are responsible for secreting exosomes after fusing with the plasma membrane[11][12][13]. The biogenesis and release of exosomes are different from microvesicle formation because microvesicles are formed by the budding of the plasma membrane whereas exosomes are released by the fusion of MVEs to the plasma membrane[1][14]. It has been found that the biogenesis and subsequent release of exosomes are fairly dependent on the physiological state and conditions of both the originating cells as well as target cells, therefore, different exosomes have varying protein, lipid, and nucleic acid profiles[1] The biogenesis of exosomes is initiated when cargoes are secreted by Golgi bodies and transported to targeted endosomal membranes which are subsequently matured into MVEs with an average diameter between 250–1000 nm. Several ILVs (30–150 nm) are formed in the lumen of MVEs during its maturation process by inward budding of the endosomal membrane of MVEs[12]. During this invagination, designated cargoes are sorted and segregated, and are further incorporated into the forming ILVs. The highly selective cargo segregation and sorting require specialized sorting pathways including the endosomal sorting complex required for transport (ESCRT)-dependent, and ESCRT-independent pathways[15][16][17]. ESCRT is an evolutionarily conserved protein complexes family, having ESCRT-0, ESCRT-I, ESCRT-II, and ESCRT-III proteins, which involves in cargo sorting and membrane forming of MVEs and ILVs[17]. The ESCRT-0 and ESCRT-I proteins segregate and associate ubiquitylated cargoes to lipid microdomains of the membrane of MVEs, followed by invagination and formation of MVEs and ILVs by ESCRT-II and ESCRT-III protein complexes. Some other proteins are also involved in the ESCRT machinery which includes ALG-2 interacting protein X (ALIX), tumor susceptibility gene 101 (TSG101), and vacuolar protein sorting-associated protein (VPS4). In the ESCRT independent mechanism, tetraspanins (CD63, CD81, CD82, CD37, and CD9) and chaperones (HSP60, HSP70, and HSP90) play a major role by clustering cargoes to lipid microdomains and successive formation of MVEs and ILVs[11][13][18]. Tetraspanins CD63 and CD81 are highly enriched on the membranes of ILVs and thus regarded as housekeeping markers for exosomes[18][19]. Syntenin is also involved in the ESCRT mechanism and has a role in the recycling and sorting of cargoes. It has been reported that both ESCRT dependent and independent mechanisms work in the biogenesis of exosomes, however, the selection of mechanism is highly dependent on their respective cargoes and cell types[13]. Energetically, the transfer and docking of newly formed MVEs with the plasma membrane are dependent on Ras associated binding (Rab) family of GTPases and the fusion of the MVEs to the plasma membrane requires soluble NSF-attachment protein receptor (SNARE) protein complexes[15][20][21]. After fusion to the plasma membrane, MVEs release the ILVs, which are termed exosomes upon release from the cell to the extracellular regions. Not all matured MVEs are transferred to plasma membranes, some of them are also directed to the lysosomal pathway for degradation[15][20].
The composition of exosomes is diverse and may reflect its origin cells or tissues. Their content includes proteins, lipids, enzymes, and nucleic acids, which play an important role in cell to cell communications and are responsible for delivering various signal molecules to both proximal and distant locations. Some of the most common cargoes on exosomes are transmembrane proteins, cytoskeletal proteins, and heat shock proteins; various lipids; and various types of RNAs e.g., mRNA, microRNA (miRNA), non-coding RNA (ncRNA), mitochondrial DNA (mtDNA), and single-stranded and double-stranded DNAs (ssDNA and dsDNA)[1][4]. Some comprehensive databases of exosomes like ExoCarta[22], Vesiclepedia[13] and EVpedia[14] are also available which have detailed information on exosomal components including proteins, lipids, and nucleic acids as well as included different methodologies of exosomes isolation and characterization. Exosomes contain certain conserved proteins that include proteins involved in ESCRT dependent exosomal biogenesis such as ALIX and TSG101, and ESCRT independent tetraspanin family of proteins like CD63, CD9, CD37, CD81, and CD82[1][14][23]. Since these proteins are absent on other types of vesicles, these can be considered as “hallmark exosomal markers”. Tetraspanins are categorized into a class of transmembrane proteins that interact with other proteins like integrins and thereby result in the transport and fusion of exosomes and helps to establish a connection with target cells[15][18][19]. Additionally, Rab GTPases, annexins, and flotillin assist in the efficient transport and fusion of exosomes[24][25]. Another important protein, syntenin, is involved in the clustering of exosomal proteins to transmembrane domains, especially CD63. Higher expression of syntenin is correlated with CD63 enrichment onto the surface of exosomes[18][19].
The exosomes are generously enriched in lipids, especially lipid rafts such as ceramides, sphingomyelin, cholesterol, sphingolipids, glycerophospholipids, and glycosphingolipids. The exosomal membrane has lysophosphatidic acid which plays an important role in the formation of ILVs from MVEs. Some lipids are found in lesser quantities which include phosphatidylserines, phosphatidylcholine, and phosphatidylinositols. Exosomes also have prostaglandins and some enzymes such as phosphatases, glycosidases, lipases, and proteases. The enzymes found in exosomes generally represent exosomal origin cell types and their metabolic activity[26].
The exosomes also contain nucleic acids that play an important role in intracellular communications as well as in the pathophysiology of a variety of diseases. Both RNA and DNA are found as exosomal cargo and their concentration and composition are dependent on the origin and the target cells[27][28][29]. Exosomal miRNAs play a significant role in intracellular communications and gene regulation. The miRNAs have been discussed in several studies due to their potency as a diagnostic marker and in the understanding of disease progression and pathology[30]. Michael et al. [30] successfully showed the isolation and characterization of salivary exosomal miRNAs (miR-31, miR-17-92, miR-125a, and miR-200a) in an easy and non-invasive way. Some studies also reported mtDNA, ssDNA and dsDNA, and genomic DNA in exosomes from various sources, however, their detailed roles and sorting mechanism are still a topic of active research and most details are yet to be elucidated[27][28].
2. Exosomal Biomarkers and Their Role in Neurodegenerative Diseases
2.1. Alzheimer’s Disease
Dementia is an umbrella term that is broadly used for the loss of cognitive functioning and memory. Medically, it can be referred to as chronic brain dysfunction. According to the recent statistical reports, the prevalence of dementia is thought to increase from 3% (age group 70–75 years) to 20–25% among those with the age approaching 85 years. It is predicted, the number of individuals currently suffering will be doubled every 20 years and may account for 81.1 million diseased people by 2040. The developing countries are among the highest sufferers (60% of all the global dementia cases in 2001, expected to rise to 71% by 2040)[33]. Although, in aged individuals, memory loss is not uncommon, the effect on one or more domains of cognition within the brain resulting in an altered social behavior is considered as a major characteristic of dementia[34]. The majority of dementia cases are dictated by AD pathology. Two-third of individuals suffering from dementia as observed in population studies have Alzheimer’s disease[35]. Therefore, on the whole, AD accounts for 70% of dementia cases[36]. The histopathological changes occurring in AD brain can be divided into two processes: Firstly, the formation of extracellular plaques (senile plaques) by deposition of amyloid-beta and secondly, the intercellular tangles/neurofibrillary tangles (NFT’s) originating from paired helical filament (PHF) of microtubule-associated protein tau[37] (Figure 1). In a normal aging brain, the presence of plaques is reported, but the neurotoxic amyloid-beta (Aβ) species forming plaques are responsible for the disease pathology [38]. The aggregates of amyloid-beta found extracellularly are formed due to the deteriorative defect in the amyloid precursor protein (APP) machinery[39]. In the most common cases, the proteolytic cleavage of the transmembrane glycoprotein APP by the help of β, γ (presenilin-1, presenilin-2) secretases aids in the formation of various Aβ species [40][41][42][43][44]. The most prominent reason for defective APP machinery that leads to aggregative nature Aβ is the defective substrate i.e., APP, or the presenilin enzyme (PSEN), which is sufficient to cause the disease[45]. The genetic mutation in the PSEN gene as well as the presence of apolipoprotein ε4 genotype makes an individual more prone to cognitive impairment and AD[46][47][48][49][50]. The formation of plaque is known to be caused by Aβ42, which is crowned as the culprit to cause disease and also portrays fibrillation inducing capabilities. Evidence suggests that polymerization of Aβ is a complex process linked with various metastable intermediaries and thus is termed as nucleated conformational conversion[51]. The elusive behavior of Aβ oligomer in the causation of AD is not very prominently known, but the present evidence suggests that the soluble oligomeric species derived from the brain strongly correlates with the decline in cognition better in comparison to plaques[52]. The soluble oligomer toxicity machinery works by three different molecular pathways[53]. Therefore, the perturbations in the maintenance of Aβ homeostasis in between the CNS and peripheral system lead to the accumulation of toxic species.
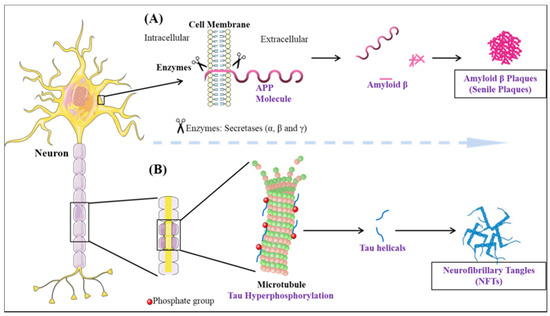
Figure 1. The neuropathological hallmarks of Alzheimer’s disease: (A) formation of amyloid-beta plaques (B) formation of neurofibrillary tangles.
The role of exosomes in the cell to cell communication and pathology of AD is beginning to unfold[54]. The exosomal protein cargos containing APP, Aβ, and tau facilitates intercellular communication and leads to further propagation of Aβ and tau pathologies[55]. In the case of AD, the loss in function of the endosomal-lysosomal system due to a heterozygous or homozygous Apo E4 genotype is one of the prime reasons for an increased exosome production [56][57][58]. The proteasomal and lysosomal system dysfunction is common in AD and is the reason why the APP containing MVE’s fuses with the plasma membrane. Generally, the late endosome can either fuse with a lysosome, resulting in the digestion of their inherent material, in presence of hydrolases, or the fusion of MVE’s consisting of ILV’s with plasma membrane which results in the formation of exosome[59][60]. Exosome shows ambiguity in its role with its, either, neuroprotective or neurodegenerative nature. In AD, the extracellular vesicles have been proven to participate in the dispersing of Aβ and thereby exuberating Aβ pathology[61][62][63][64]. A similar exosomal dependent spread of hyperphosphorylated tau is also reported[65][66][67][68]. It is also observed that EVs contain APP, C-terminal fragments of APP, and the various isoforms of Aβ.
The exosomal cargo content including proteins, lipids, and nucleic acids are gaining importance as prospective biomarkers. In the case of diseases like AD, where behavioral symptoms occur much later in life than the actual events of disease progression, the search for identification of early screening markers has been of utmost importance. Within CSF derived exosomes from severe AD patients, the decreased expression levels of Aβ were reported[69]. In another study from plasma-derived exosomes, Aβ was higher and their level was lower when compared to exosomes derived from other sources[70]. The level of soluble Aβ42 and other proteins involved in the Aβ42 generating pathway is higher within the astrocytic derived exosomes in comparison to the neuronal exosomes [71]. The ratio of p-tau/total tau also increases in the case of AD[72]. The activity of lactoferrin and acetylcholinesterase was also assessed in AD patients[73][74].
2.2. Parkinson’s Disease
Parkinson’s disease is the second most common neurodegenerative disease after AD[75]. It is a movement disorder of older age with 2–3% of > 65 years being affected[76]. The incidence rate of PD ranges from 5 to > 35 newer cases per 100,000 individuals worldwide[77]. The onset of behavioral symptoms of PD is rare before 50 years of age [77][78]. The occurrence of this disease is thought to be more common in men than in women[79], with an exception of the study completed in Japan that concluded the prevalence is gender unbiased[80]. The men’s susceptibility to the disease has led to the notion that women’s sex hormones may portray a protective mechanism against this disease, however, this view is still debatable[81]. Parkinson’s disease is a prion-based neurodegenerative disorder [82], which is known to be associated with the loss of dopaminergic neurons in the substantia nigra pars compacta portion of the midbrain and the prime histopathological change i.e., lesions are seen within this region. Majorly the lesions are in the ventrolateral portion of substantia nigra[83][84]. The appearance of lesions is due to the depigmenting of the dopaminergic neurons[85]. The key molecular and neurophysiological mechanism of PD pathology is the intraneuronal aggregation of the misfolded alpha-synuclein protein and the presence of Lewy bodies (Figure 2). The presence of Lewy bodies is the prime neuropathological change accompanying aggregation of oligomeric alpha-synuclein (α-syn)[86]. α-syn acts as a molecular chaperone and has a role in intracellular trafficking and synaptic vesicle transportation[87][88]. α-syn is required for the release of neurotransmitters as it facilitates the association of synaptic vesicles with SNAREs assembly[89]. The other accomplice of PD pathology, the Lewy bodies, which spread from the serotonergic and cholinergic neurons to the limbic and the neocortex regions[90]. A Parkinson’s affected individual shows both motor and non-motor symptoms. The major motor symptoms are bradykinesia, resting tremor, postural instability, stooped structure, freezing, or rigidity. The most significant non-motor symptoms are hyposmia, rapid-eye ball movement sleep disorder (RBD), constipation, urinary dysfunction, depression, and hallucinations accompanied by impairment in cognition[91][92][93][94]. The spread of PD occurs in the Braak staging manner[95]. The symptoms of PD can be easily managed by the currently available treatments. The highly efficacious therapies which use the L-dopa and the deep brain stimulation have made PD the only neurodegenerative disease with manageable symptoms[96][97][98][99]. The mechanisms of aggregated α-syn neurotoxicity include mitochondrial defects, proteasomal effects, endoplasmic reticulum stress, and inflammatory responses[100]. The oligomeric α-syn results in a multitude of conditions in mitochondria and endoplasmic reticulum: Mitotoxicity by a decrement in calcium retention time and increasing cytochrome c release[101]. The transgenic mouse model with an A53T point mutation of the SNCA gene results in deleterious effects on the ER protein quality due to the overproduction of α-syn oligomers [102].
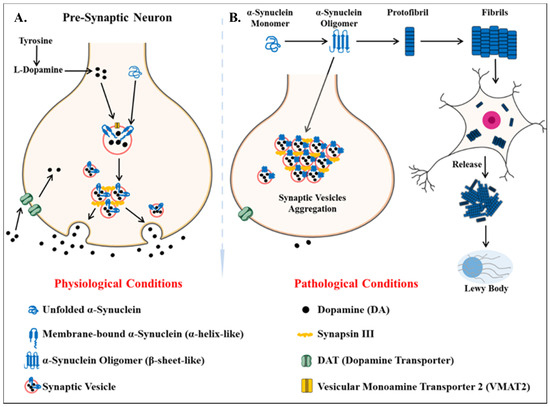
Figure 2. Key differentiation in the normal physiological state and pathological state of dopamine metabolism and homeostasis of alpha-synuclein protein (α -syn) (A) the normal physiological state: Homeostasis of alpha-synuclein protein maintained (B) the perturbations in alpha-synuclein homeostasis as well interneuronal aggregation of α-syn oligomer and formation of lewy bodies.
The genetically linked familial form of PD is associated with the point mutation in the SNCA gene, encoding the α-syn protein[103]. The perturbations in proteostasis and degradation of α-syn are cardinal to the development of PD pathology[100][104]. Generally, α-syn is present in the monomeric form, but after the acquisition of neurotoxic properties, it undergoes oligomerization and further aggregates to form protofibrils[105]. The degradation of α-syn occurs by the lysosomal autophagy system (LAS) and proteasomal pathway[104]. The LAS pathway has mainly been associated with the clearance of α-syn oligomeric assemblies [104][106].
The role of exosomes in PD results in the seeding of intraneuronal α-syn in a prion-like manner. Exosomal machinery serves as an auxiliary mechanism for dissipating early molecular changes of PD pathology to other cells[107]. The genetic mutations occurring in PARK-LRRK2′s gene locus 12q12 are linked with the faulty LAS machinery[108]. The G2019S mutation in LRRK2 is associated with the impaired functioning of the LAS pathway, thus corroborating with the disturbed homeostasis of α-syn leading to more aggregated form and further results in depigmentation of dopaminergic neurons. The α-syn containing MVB’s are formed from α-syn possessing endosomes and thus aid in the transmission of PD pathology after fusion with the plasma membrane[109][110]. It is also reported that the release of exosomal associated α-syn is regulated by the intracellular concentration of calcium[111]. The ambient environment of exosomes is also suggested to promote aggregation of α-syn and aid in spreading the PD pathology[112]. It was observed that α-syn increases the secretion of exosomes by microglia BV-2 cells of mice[113]. Moreover, the inhibition of the LAS pathway has shown an increased production of exosomal cargo with α-syn and decreased intra-neuronal α-syn aggregation, thus establishing the neuroprotective role of exosomes[111]. An experimental observation suggests that R1441C LRRK2 mutation aids in the induction of large MVB’s and thereby facilitating the exosome release.
In recent years, the search for CSF and plasma biomarkers in PD has been of prime importance for the diagnosis of disease pathology or the associated neuropathological change in an early stage[114][115]. The study on astrocytic and oligodendrocyte derived exosomes from plasma of PD patients showed a high increase in early-stage PD patients and the concentration of exosomes corroborated with the disease severity[116]. Another study performed where the protein profiling of the plasma-derived exosomes from PD patients at Hoehn and Yahr (HY) stages one, two, and three was done suggested apolipoprotein A1 can be a potential biomarker to monitor disease progression of PD [117]. The level of the α-syn oligomer, α-syn oligomer/α-syn total in salivary exosomes were found to be higher in PD patients but did not correlate with the disease severity[118]. The level of protein DJ1 was observed to be higher in PD patients[119]. Some key studies elaborating on the role of exosomes in the early diagnosis of neurodegenerative diseases are tabulated in Table 2.
Table 2. Studies describing the potential role of exosomes as an early diagnostics for neurodegenerative diseases. (“__” signifies no studies are reported in this subcategory).
| Neurodegenerative Disease | Source of Exosome | Studied Exosome Cargo Content | References |
|---|---|---|---|
| Alzheimer’s disease | Saliva |
|
[120] |
| CSF |
|
[121][122][123][124][125][126][127][128] | |
| Plasma |
|
[129][130][131][132][133][134][135][136] | |
| Serum |
|
[137] | |
| Urine |
|
[138][139] | |
| Parkinson’s disease | Saliva |
|
[118][140] |
| CSF |
|
[64][141][142] | |
| Plasma |
|
[116][143][144][145][146][147] | |
| Urine |
|
[148][149] | |
| Huntington Disease | Saliva | ___ | ___ |
| CSF |
|
[150] | |
| Plasma |
|
[151][152][153] | |
| Urine | ____ | ___ | |
| Amyotrophic Lateral Sclerosis | Saliva | ____ | ___ |
| CSF |
|
[154][155][156] | |
| Plasma |
|
[157] | |
| Urine | ____ | ____ |
2.3. Other Neurodegenerative Disorders
The two additional, but less prevalent neurodegenerative diseases are Huntington’s disease and amyotrophic lateral sclerosis. HD is linked primarily to the expanded trinucleotide repeat in the huntingtin gene (HTT) which is the pathological carrier-a mutant form of the multi-functional protein huntingtin. ALS is linked to the copper-zinc superoxide dismutase 1 (SOD1) gene mutation and protein inclusions are ubiquitinated and enriched in tar DNA binding protein-43 (TDP-43) and has concomitant behavioral symptoms like FTD. Huntington is another category of prion-based neurodegenerative disease which is autosomal dominant. It is known to panoply the motor, cognitive and behavioral defects. The abnormal amplification of CAG repeats in the Huntington gene is the accountable cause of this disease[158][159]. The mechanism which regulates the production of mutant huntingtin protein (mHTT) still appears translucent. The prevalence rate of HD is 10.6–13.7 individuals per 100,000[160][161]. The transmission of mHTT between neurons occurs by tunneling nanotubes and/or vesicle mechanism[125]. There are not many studies that are suggestive of the role of exosomes in the transmission of mHTT. A study on 239T cells, overexpressing HTT-exon1polyQ-GFP reports the presence of polyQGFP and amplified duplicate RNA in EVs. The uptake of EVs by neurons is also observed with an increased presence of polyQ-GFP RNA, although it does not result in further toxicity[125][162]. Another study performed on plasma-derived exosomes of HD patients revealed the presence of 13 miRNAs, which are significantly upregulated[152]. The therapeutic role of exosomes from astrocytic stem cells (ASC-exo) in the in vitro model of HD, the result shows decreased production of the aggregate of mHTT of HD[162]. The presence of mHTT is not seen in astrocytes, although it was observed the increased exosome secretion from astrocytes in HD140Q knock-in (KI) mice. The N-terminal mHTT accumulates in the nuclei and forms aggregates, resulting in the secretion of exosomes from cultured astrocytes[164]. The level of total huntingtin protein in the saliva is higher in HD patients when compared to healthy cohorts[165].
Amyotrophic lateral sclerosis comes under the umbrella of prion-based neurodegenerative disorders[166]. ALS is a motor neuron disease that focally spreads in lower and upper motor neurons. It spreads in the motor neuron of the motor cortex, brainstem, and spinal cord[167]. The progression of this disease varies from 3-5 years to extreme slow progression in some affected individuals[167]. The disease shows diverse symptoms ranging from early-onset in the spinal cord characterized by muscle weakness in lower limbs to the bulbar-onset characterized by dysarthria and dysphagia[168]. The impairment in cognition is an accomplice to the cardinal symptoms of ALS[168]. The global prevalence rates of ALS are 5 affected people amongst 100,000[167]. The most significant genetic link of ALS has persisting connections with the SOD1 gene, which encodes the superoxide-dismutase protein[169]. SOD1 is responsible for the conversion of super-oxide molecules to hydrogen peroxide. There are more than 30 mutations on different genes present amongst which only 14 mutations give rise to the casualty in disease[170][171][172][173][174]. These mutations are involved in protein control, cytoskeletal dynamics, RNA processing, and metabolism, and proteostasis. The diverse mutations recognized in ALS are similar to those accountable for symptoms development in FTD. The presence of C9orf72, an aberrant hexanucleotide (GGGGCC) expansion in the non-coding regions of ALS patients is in similarity to the patients of FTD which characterizes ALS as a neurodegenerative disorder [173][175]. The role of extracellular vesicles in the transmission of ALS pathology in a prion-like manner starts with the accumulation and transport of mutant proteins onto EVs which further aids in transmission across the brain cells [176]. The experimental observations have shown the reduced interaction of C9orf72 and Rab7L1 which are accountable for regulation of a fusion of MVE’s with the plasma membrane[176][177]. SOD1 was the first ALS-associated protein found in EV’s of stable mouse motor neuron-like cells having overexpressed human wild-type and mutant SOD1[178]. TDP-43, which is known to be the pathological hallmark of ALS, is identified in the brain-tissue isolated exosomes from ALS patients[179]. The secretion of exosomes is also shown to be inhibited with the knocked-out GW4869 OR RAB27A gene, responsible for TDP-43 aggregation[180].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22010440
References
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514.
- Ratajczak, M.Z.; Ratajczak, J. Extracellular microvesicles/exosomes: Discovery, disbelief, acceptance, and the future? Leukemia 2020.
- Chung, I.-M.; Rajakumar, G.; Venkidasamy, B.; Subramanian, U.; Thiruvengadam, M. Exosomes: Current use and future applications. Clin. Chim. Acta 2020, 500, 226–232.
- Cheshmi, B.; Cheshomi, H. Salivary exosomes: Properties, medical applications, and isolation methods. Mol. Biol. Rep. 2020, 47, 6295–6307.
- Conigliaro, A.; Corrado, C.; Fontana, S.; Alessandro, R. Chapter 1—Exosome basic mechanisms. In Exosomes; Edelstein, L., Smythies, J., Quesenberry, P., Noble, D., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 1–21.
- Trams, E.G.; Lauter, C.J.; Norman, S., Jr.; Heine, U. Exfoliation of membrane ecto-enzymes in the form of micro-vesicles. Biochim. Biophys. Acta (BBA) Biomembr. 1981, 645, 63–70.
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420.
- Greening, D.W.; Xu, R.; Ji, H.; Tauro, B.J.; Simpson, R.J. A protocol for exosome isolation and characterization: Evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. Methods Mol. Biol. 2015, 1295, 179–209.
- Sokolova, V.; Ludwig, A.K.; Hornung, S.; Rotan, O.; Horn, P.A.; Epple, M.; Giebel, B. Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids and surfaces. Colloids Surf. B Biointerfaces 2011, 87, 146–150.
- van der Pol, E.; Coumans, F.A.; Grootemaat, A.E.; Gardiner, C.; Sargent, I.L.; Harrison, P.; Sturk, A.; van Leeuwen, T.G.; Nieuwland, R. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J. Thromb. Haemost. 2014, 12, 1182–1192.
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289.
- Heijnen, H.F.G.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated Platelets Release Two Types of Membrane Vesicles: Microvesicles by Surface Shedding and Exosomes Derived From Exocytosis of Multivesicular Bodies and alpha-Granules. Blood 1999, 94, 3791–3799.
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641.
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228.
- Stuffers, S.; Sem Wegner, C.; Stenmark, H.; Brech, A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic 2009, 10, 925–937.
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT Pathway. Dev. Cell 2011, 21, 77–91.
- Radulovic, M.; Stenmark, H. ESCRTs in membrane sealing. Biochem. Soc. Trans. 2018, 46, 773–778.
- Lötvall, J.; Hill, A.F.; Hochberg, F.; Buzás, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913.
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968.
- Klumperman, J.; Raposo, G. The complex ultrastructure of the endolysosomal system. Cold Spring Harbor Perspect. Biol. 2014, 6, a016857.
- Anand, S.; Samuel, M.; Kumar, S.; Mathivanan, S. Ticket to a bubble ride: Cargo sorting into exosomes and extracellular vesicles. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2019, 1867, 140203.
- Mathivanan, S.; Simpson, R.J. ExoCarta: A compendium of exosomal proteins and RNA. Proteomics 2009, 9, 4997–5000.
- Hemler, M.E. Tetraspanin Proteins Mediate Cellular Penetration, Invasion, and Fusion Events and Define a Novel Type of Membrane Microdomain. Annu. Rev. Cell Dev. Biol. 2003, 19, 397–422.
- Hsu, C.; Morohashi, Y.; Yoshimura, S.-i.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Grønborg, M.; Möbius, W.; Rhee, J.; et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A–C. J. Cell Biol. 2010, 189, 223–232.
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30.
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41.
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769.
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075.
- Fan, Q.; Yang, L.; Zhang, X.; Peng, X.; Wei, S.; Su, D.; Zhai, Z.; Hua, X.; Li, H. The emerging role of exosome-derived non-coding RNAs in cancer biology. Cancer Lett. 2018, 414, 107–115.
- Michael, A.; Bajracharya, S.D.; Yuen, P.S.T.; Zhou, H.; Star, R.A.; Illei, G.G.; Alevizos, I. Exosomes from human saliva as a source of microRNA biomarkers. Oral Dis. 2010, 16, 34–38.
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117.
- Elahi, F.M.; Miller, B.L. A clinicopathological approach to the diagnosis of dementia. Nat. Rev. Neurol. 2017, 13, 457–476.
- Morley, J.E.; Morris, J.C.; Berg-Weger, M.; Borson, S.; Carpenter, B.D.; Del Campo, N.; Dubois, B.; Fargo, K.; Fitten, L.J.; Flaherty, J.H.; et al. Brain health: The importance of recognizing cognitive impairment: An IAGG consensus conference. J. Am. Med. Dir. Assoc. 2015, 16, 731–739.
- van der Flier, W.M.; Scheltens, P. Epidemiology and risk factors of dementia. J. Neurol. Neurosurg. Psychiatry 2005, 76 (Suppl. 5), v2–v7.
- Perl, D.P. Neuropathology of Alzheimer’s disease. Mt. Sinai J. Med. 2010, 77, 32–42.
- Kumar, S.; Rezaei-Ghaleh, N.; Terwel, D.; Thal, D.R.; Richard, M.; Hoch, M.; Mc Donald, J.M.; Wüllner, U.; Glebov, K.; Heneka, M.T. Extracellular phosphorylation of the amyloid β-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. EMBO J. 2011, 30, 2255–2265.
- Goate, A.; Chartier-Harlin, M.-C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706.
- Kang, J.; Lemaire, H.; Unterbeck, A.; Salbaum, J.; Masters, C.; Grzeschik, K.; Multihaup, G.; Beyreuther, K.; Muller-Hill, B. Amyloid production secretase. Nature 1987, 325, 733–736.
- Weidemann, A.; König, G.; Bunke, D.; Fischer, P.; Salbaum, J.; Masters, C.L.; Beyreuther, K. Cell 1989, 57, 115–126.
- Sisodia, S.; Koo, E.; Beyreuther, K.; Unterbeck, A.; Price, D. Evidence that beta-amyloid protein in Alzheimer’s disease is not derived by normal processing. Science 1990, 248, 492–495.
- Esch, F.S.; Keim, P.S.; Beattie, E.C.; Blacher, R.W.; Culwell, A.R.; Oltersdorf, T.; McClure, D.; Ward, P.J. Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science 1990, 248, 1122–1124.
- De Strooper, B. Proteases and proteolysis in Alzheimer disease: A multifactorial view on the disease process. Physiol. Rev. 2010, 90, 465–494.
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099.
- Chartier-Hariln, M.-C.; Parfitt, M.; Legrain, S.; Pérez-Tur, J.; Brousseau, T.; Evans, A.; Berr, C.; Vldal, O.; Roques, P.; Gourlet, V. Apolipoprotein E, ɛ4 allele as a major risk factor for sporadic early and late-onset forms of Alzheimer’s disease: Analysis of the 19q13. 2 chromosomal region. Hum. Mol. Genet. 1994, 3, 569–574.
- Houlden, H.; Crook, R.; Backhovens, H.; Prihar, G.; Baker, M.; Hutton, M.; Rossor, M.; Martin, J.J.; Van Broeckhoven, C.; Hardy, J. ApoE genotype is a risk factor in nonpresenilin early-onset alzheimer’s disease families. Am. J. Med. Genet. 1998, 81, 117–121.
- Rebeck, G.W.; Reiter, J.S.; Strickland, D.K.; Hyman, B.T. Apolipoprotein E in sporadic Alzheimer’s disease: Allelic variation and receptor interactions. Neuron 1993, 11, 575–580.
- Shi, J.; Han, P.; Kuniyoshi, S.M. Cognitive impairment in neurological diseases: Lessons from apolipoprotein E. J. Alzheimer’s Dis. 2014, 38, 1–9.
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118.
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T., Jr. The carboxy terminus of the. beta. amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32, 4693–4697.
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Vbeyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866.
- Mc Donald, J.M.; Savva, G.M.; Brayne, C.; Welzel, A.T.; Forster, G.; Shankar, G.M.; Selkoe, D.J.; Ince, P.G.; Walsh, D.M. The presence of sodium dodecyl sulphate-stable Aβ dimers is strongly associated with Alzheimer-type dementia. Brain 2010, 133, 1328–1341.
- Samanta, S.; Rajasingh, S.; Drosos, N.; Zhou, Z.; Dawn, B.; Rajasingh, J. Exosomes: New molecular targets of diseases. Acta Pharmacol. Sin. 2018, 39, 501–513.
- D’anca, M.; Fenoglio, C.; Serpente, M.; Arosio, B.; Cesari, M.; Scarpini, E.A.; Galimberti, D. Exosome determinants of physiological aging and age related neurodegenerative diseases. Front. Aging Neurosci. 2019, 11, 232.
- Raber, J.; Wong, D.; Buttini, M.; Orth, M.; Bellosta, S.; Pitas, R.E.; Mahley, R.W.; Mucke, L. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: Increased susceptibility of females. Proc. Natl. Acad. Sci. USA 1998, 95, 10914–10919.
- Holtzman, D.M.; Bales, K.R.; Tenkova, T.; Fagan, A.M.; Parsadanian, M.; Sartorius, L.J.; Mackey, B.; Olney, J.; McKeel, D.; Wozniak, D. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2892–2897.
- Caselli, R.J.; Dueck, A.C.; Osborne, D.; Sabbagh, M.N.; Connor, D.J.; Ahern, G.L.; Baxter, L.C.; Rapcsak, S.Z.; Shi, J.; Woodruff, B.K. Longitudinal modeling of age-related memory decline and the APOE ε4 effect. N. Engl. J. Med. 2009, 361, 255–263.
- Selwood, S.P.; Parvathy, S.; Cordell, B.; Ryan, H.S.; Oshidari, F.; Vincent, V.; Yesavage, J.; Lazzeroni, L.C.; Murphy, G.M., Jr. Gene expression profile of the PDAPP mouse model for Alzheimer’s disease with and without Apolipoprotein, E. Neurobiol. Aging 2009, 30, 574–590.
- Trommer, B.L.; Shah, C.; Yun, S.H.; Gamkrelidze, G.; Pasternak, E.S.; Ye, G.L.; Sotak, M.; Sullivan, P.M.; Pasternak, J.F.; LaDu, M.J. ApoE isoform affects LTP in human targeted replacement mice. Neuroreport 2004, 15, 2655–2658.
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease β-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177.
- Sharples, R.A.; Vella, L.J.; Nisbet, R.M.; Naylor, R.; Perez, K.; Barnham, K.J.; Masters, C.L.; Hill, A.F. Inhibition of γ-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB J. 2008, 22, 1469–1478.
- Morales, R.; Duran-Aniotz, C.; Castilla, J.; Estrada, L.; Soto, C. De novo induction of amyloid-β deposition in vivo. Mol. Psychiatry 2012, 17, 1347–1353.
- Joshi, P.; Benussi, L.; Furlan, R.; Ghidoni, R.; Verderio, C. Extracellular vesicles in Alzheimer’s disease: Friends or foes? Focus on aβ-vesicle interaction. Int. J. Mol. Sci. 2015, 16, 4800–4813.
- Ghate, P.S.; Sidhar, H.; Carlson, G.A.; Giri, R.K. Development of a novel cellular model of Alzheimer’s disease utilizing neurosphere cultures derived from B6C3-Tg(APPswe, PSEN1dE9)85Dbo/J embryonic mouse brain. SpringerPlus 2014, 3, 161.
- Koo, E.H.; Squazzo, S.L. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 1994, 269, 17386–17389.
- Shankar, G.M.; Leissring, M.A.; Adame, A.; Sun, X.; Spooner, E.; Masliah, E.; Selkoe, D.J.; Lemere, C.A.; Walsh, D.M. Biochemical and immunohistochemical analysis of an Alzheimer’s disease mouse model reveals the presence of multiple cerebral Aβ assembly forms throughout life. Neurobiol. Dis. 2009, 36, 293–302.
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006, 440, 352–357.
- Simonsen, A.H.; Herukka, S.-K.; Andreasen, N.; Baldeiras, I.; Bjerke, M.; Blennow, K.; Engelborghs, S.; Frisoni, G.B.; Gabryelewicz, T.; Galluzzi, S. Recommendations for CSF AD biomarkers in the diagnostic evaluation of dementia. Alzheimer’s Dement. 2017, 13, 274–284.
- Hornung, S.; Dutta, S.; Bitan, G. CNS-Derived Blood Exosomes as a Promising Source of Biomarkers: Opportunities and Challenges. Front. Mol. Neurosci. 2020, 13, 38.
- Goetzl, E.J.; Mustapic, M.; Kapogiannis, D.; Eitan, E.; Lobach, I.V.; Goetzl, L.; Schwartz, J.B.; Miller, B.L. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. Faseb J. 2016, 30, 3853–3859.
- Shi, M.; Sui, Y.-T.; Peskind, E.R.; Li, G.; Hwang, H.; Devic, I.; Ginghina, C.; Edgar, J.S.; Pan, C.; Goodlett, D.R.; et al. Salivary Tau Species are Potential Biomarkers of Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 27, 299–305.
- Bermejo-Pareja, F.; del Ser, T.; Valentí, M.; de la Fuente, M.; Bartolome, F.; Carro, E. Salivary lactoferrin as biomarker for Alzheimer’s disease: Brain-immunity interactions. Alzheimer’s Dement. 2020, 16, 1196–1204.
- Ahmadi-Motamayel, F.; Goodarzi, M.T.; Tarazi, S.; Vahabian, M. Evaluation of salivary acetylcholinesterase and pseudocholinesterase in patients with Alzheimer’s disease: A case–control study. Spec. Care Dent. 2019, 39, 39–44.
- Lebouvier, T.; Chaumette, T.; Paillusson, S.; Duyckaerts, C.; Bruley des Varannes, S.; Neunlist, M.; Derkinderen, P. The second brain and Parkinson’s disease. Eur. J. Neurosci. 2009, 30, 735–741.
- Poewe, W.; Seppi, K.; Tanner, C.; Halliday, G.; Brundin, P.; Volkmann, J.; Schrag, A.; Lang, A. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013.
- Twelves, D.; Perkins, K.S.; Counsell, C. Systematic review of incidence studies of Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2003, 18, 19–31.
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2014, 29, 1583–1590.
- Baldereschi, M.; Di Carlo, A.; Rocca, W.; Vanni, P.; Maggi, S.; Perissinotto, E.; Grigoletto, F.; Amaducci, L.; Inzitari, D. Parkinson’s disease and parkinsonism in a longitudinal study: Two-fold higher incidence in men. Neurology 2000, 55, 1358–1363.
- Kusumi, M.; Nakashima, K.; Harada, H.; Nakayama, H.; Takahashi, K. Epidemiology of Parkinson’s disease in Yonago City, Japan: Comparison with a study carried out 12 years ago. Neuroepidemiology 1996, 15, 201–207.
- Cerri, S.; Mus, L.; Blandini, F. Parkinson’s Disease in Women and Men: What’s the Difference? J. Parkinson’s Dis. 2019, 9, 501–515.
- Ma, J.; Gao, J.; Wang, J.; Xie, A. Prion-like mechanisms in Parkinson’s disease. Front. Neurosci. 2019, 13, 552.
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain 1991, 114, 2283–2301.
- Damier, P.; Hirsch, E.; Agid, Y.; Graybiel, A. The substantia nigra of the human brain: II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122, 1437–1448.
- Dickson, D.W. Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harbor Perspect. Med. 2012, 2, a009258.
- Irizarry, M.C.; Growdon, W.; Gomez-Isla, T.; Newell, K.; George, J.M.; Clayton, D.F.; Hyman, B.T. Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson’s disease and cortical Lewy body disease contain α-synuclein immunoreactivity. J. Neuropathol. Exp. Neurol. 1998, 57, 334–337.
- Vekrellis, K.; Xilouri, M.; Emmanouilidou, E.; Rideout, H.J.; Stefanis, L. Pathological roles of α-synuclein in neurological disorders. Lancet Neurol. 2011, 10, 1015–1025.
- Burré, J. The synaptic function of α-synuclein. J. Parkinson’s Dis. 2015, 5, 699–713.
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472.
- Halliday, G.M.; McCann, H. The progression of pathology in Parkinson’s disease. Ann. N. Y. Acad. Sci. 2010, 1184, 188–195.
- Tolosa, E.; Wenning, G.; Poewe, W. The diagnosis of Parkinson’s disease. Lancet Neurol. 2006, 5, 75–86.
- Chaudhuri, K.R.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Dopaminergic pathophysiology and treatment. Lancet Neurol. 2009, 8, 464–474.
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601.
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912.
- Burke, R.E.; Dauer, W.T.; Vonsattel, J.P.G. A critical evaluation of the Braak staging scheme for Parkinson’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2008, 64, 485–491.
- Stayte, S.; Vissel, B. Advances in non-dopaminergic treatments for Parkinson’s disease. Front. Neurosci. 2014, 8, 113.
- Bronstein, J.M.; Tagliati, M.; Alterman, R.L.; Lozano, A.M.; Volkmann, J.; Stefani, A.; Horak, F.B.; Okun, M.S.; Foote, K.D.; Krack, P. Deep brain stimulation for Parkinson disease: An expert consensus and review of key issues. Arch. Neurol. 2011, 68, 165.
- Stern, M.B.; Follett, K.A.; Weaver, F.M. Randomized trial of deep brain stimulation for Parkinson disease: Thirty-six-month outcomes; turning tables: Should GPi become the preferred DBS target for Parkinson disease? Author response. Neurology 2013, 80, 225.
- Deuschl, G.; Agid, Y. Subthalamic neurostimulation for Parkinson’s disease with early fluctuations: Balancing the risks and benefits. Lancet Neurol. 2013, 12, 1025–1034.
- Bengoa-Vergniory, N.; Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Alpha-synuclein oligomers: A new hope. Acta Neuropathol. 2017, 134, 819–838.
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507.
- Lindström, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergström, J.; Erlandsson, A. Extensive uptake of α-synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol. Cell. Neurosci. 2017, 82, 143–156.
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047.
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Alpha-synuclein and protein degradation systems: A reciprocal relationship. Mol. Neurobiol. 2013, 47, 537–551.
- De Oliveira, G.A.; Silva, J.L. Alpha-synuclein stepwise aggregation reveals features of an early onset mutation in Parkinson’s disease. Commun. Biol. 2019, 2, 1–13.
- Brundin, P.; Li, J.-Y.; Holton, J.L.; Lindvall, O.; Revesz, T. Research in motion: The enigma of Parkinson’s disease pathology spread. Nat. Rev. Neurosci. 2008, 9, 741–745.
- Yu, H.; Sun, T.; An, J.; Wen, L.; Liu, F.; Bu, Z.; Cui, Y.; Feng, J. Potential Roles of Exosomes in Parkinson’s Disease: From Pathogenesis, Diagnosis, and Treatment to Prognosis. Front. Cell Dev. Biol. 2020.
- Paisan-Ruiz, C.; Lewis, P.A.; Singleton, A.B. LRRK2: Cause, risk, and mechanism. J. Parkinson’s Dis. 2013, 3, 85–103.
- Ben Gedalya, T.; Loeb, V.; Israeli, E.; Altschuler, Y.; Selkoe, D.J.; Sharon, R. α-Synuclein and Polyunsaturated Fatty Acids Promote Clathrin-Mediated Endocytosis and Synaptic Vesicle Recycling. Traffic 2009, 10, 218–234.
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; McIlwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking α-synuclein. J. Neurosci. 2002, 22, 8797–8807.
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851.
- Grey, M.; Dunning, C.J.; Gaspar, R.; Grey, C.; Brundin, P.; Sparr, E.; Linse, S. Acceleration of α-synuclein aggregation by exosomes. J. Biol. Chem. 2015, 290, 2969–2982.
- Chang, C.; Lang, H.; Geng, N.; Wang, J.; Li, N.; Wang, X. Exosomes of BV-2 cells induced by alpha-synuclein: Important mediator of neurodegeneration in PD. Neurosci. Lett. 2013, 548, 190–195.
- Hall, S.; Surova, Y.; Öhrfelt, A.; Zetterberg, H.; Lindqvist, D.; Hansson, O. CSF biomarkers and clinical progression of Parkinson disease. Neurology 2015, 84, 57–63.
- Chang, C.-W.; Yang, S.-Y.; Yang, C.-C.; Chang, C.-W.; Wu, Y.-R. Plasma and Serum Alpha-Synuclein as a Biomarker of Diagnosis in Patients with Parkinson’s Disease. Front. Neurol. 2020, 10, 1388.
- Ohmichi, T.; Mitsuhashi, M.; Tatebe, H.; Kasai, T.; El-Agnaf, O.M.A.; Tokuda, T. Quantification of brain-derived extracellular vesicles in plasma as a biomarker to diagnose Parkinson’s and related diseases. Parkinsonism Relat. Disord. 2019, 61, 82–87.
- Kitamura, Y.; Kojima, M.; Kurosawa, T.; Sasaki, R.; Ichihara, S.; Hiraku, Y.; Tomimoto, H.; Murata, M.; Oikawa, S. Proteomic profiling of exosomal proteins for blood-based biomarkers in Parkinson’s Disease. Neuroscience 2018, 392, 121–128.
- Cao, Z.; Wu, Y.; Liu, G.; Jiang, Y.; Wang, X.; Wang, Z.; Feng, T. α-Synuclein in salivary extracellular vesicles as a potential biomarker of Parkinson’s disease. Neurosci. Lett. 2019, 696, 114–120.
- Masters, J.M.; Noyce, A.J.; Warner, T.T.; Giovannoni, G.; Proctor, G.B. Elevated salivary protein in Parkinson’s disease and salivary DJ-1 as a potential marker of disease severity. Parkinsonism Relat. Disord. 2015, 21, 1251–1255.
- Rani, K.; Rastogi, S.; Vishwakarma, P.; Bharti, P.S.; Sharma, V.; Renu, K.; Modi, G.P.; Vishnu, V.Y.; Chatterjee, P.; Dey, A.B. A novel approach to correlate the salivary exosomes and their protein cargo in the progression of cognitive impairment into Alzheimer’s disease. J. Neurosci. Methods 2020.
- McKeever, P.M.; Schneider, R.; Taghdiri, F.; Weichert, A.; Multani, N.; Brown, R.A.; Boxer, A.L.; Karydas, A.; Miller, B.; Robertson, J. MicroRNA expression levels are altered in the cerebrospinal fluid of patients with young-onset Alzheimer’s disease. Mol. Neurobiol. 2018, 55, 8826–8841.
- Jain, G.; Stuendl, A.; Rao, P.; Berulava, T.; Centeno, T.P.; Kaurani, L.; Burkhardt, S.; Delalle, I.; Kornhuber, J.; Hüll, M. A combined miRNA–piRNA signature to detect Alzheimer’s disease. Transl. Psychiatry 2019, 9, 1–12.
- Jia, L.; Qiu, Q.; Zhang, H.; Chu, L.; Du, Y.; Zhang, J.; Zhou, C.; Liang, F.; Shi, S.; Wang, S. Concordance between the assessment of Aβ42, T-tau, and P-T181-tau in peripheral blood neuronal-derived exosomes and cerebrospinal fluid. Alzheimer’s Dement. 2019, 15, 1071–1080.
- Xiao, T.; Zhang, W.; Jiao, B.; Pan, C.-Z.; Liu, X.; Shen, L. The role of exosomes in the pathogenesis of Alzheimer’disease. Transl. Neurodegener. 2017, 6, 3.
- Wang, J.K.; Langfelder, P.; Horvath, S.; Palazzolo, M.J. Exosomes and homeostatic synaptic plasticity are linked to each other and to Huntington’s, Parkinson’s, and other neurodegenerative diseases by database-enabled analyses of comprehensively curated datasets. Front. Neurosci. 2017, 11, 149.
- Muraoka, S.; Jedrychowski, M.P.; Yanamandra, K.; Ikezu, S.; Gygi, S.P.; Ikezu, T. Proteomic Profiling of Extracellular Vesicles Derived from Cerebrospinal Fluid of Alzheimer’s Disease Patients: A Pilot Study. Cells 2020, 9, 1959.
- Liu, C.-G.; Song, J.; Zhang, Y.-Q.; Wang, P.-C. MicroRNA-193b is a regulator of amyloid precursor protein in the blood and cerebrospinal fluid derived exosomal microRNA-193b is a biomarker of Alzheimer’s disease. Mol. Med. Rep. 2014, 10, 2395–2400.
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 2015, 6, 37043.
- Goetzl, E.J.; Kapogiannis, D.; Schwartz, J.B.; Lobach, I.V.; Goetzl, L.; Abner, E.L.; Jicha, G.A.; Karydas, A.M.; Boxer, A.; Miller, B.L. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016, 30, 4141–4148.
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015, 85, 40–47.
- Winston, C.N.; Goetzl, E.J.; Schwartz, J.B.; Elahi, F.M.; Rissman, R.A. Complement protein levels in plasma astrocyte-derived exosomes are abnormal in conversion from mild cognitive impairment to Alzheimer’s disease dementia. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2019, 11, 61–66.
- Gámez-Valero, A.; Campdelacreu, J.; Vilas, D.; Ispierto, L.; Reñé, R.; Álvarez, R.; Armengol, M.P.; Borràs, F.E.; Beyer, K. Exploratory study on microRNA profiles from plasma-derived extracellular vesicles in Alzheimer’s disease and dementia with Lewy bodies. Transl. Neurodegener. 2019, 8, 31.
- Zhang, N.; Gu, D.; Meng, M.; Gordon, M.L. TDP-43 Is Elevated in Plasma Neuronal-Derived Exosomes of Patients With Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 166.
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2016, 3, 63–72.
- He, M.; Sun, L.; Cao, W.; Yin, C.; Sun, W.; Liu, P.; Tan, L.; Xu, Z.; Zhao, W. Association between plasma exosome neurogranin and brain structure in patients with Alzheimer’s disease: A protocol study. BMJ Open 2020, 10, e036990.
- Guix, F.X.; Corbett, G.T.; Cha, D.J.; Mustapic, M.; Liu, W.; Mengel, D.; Chen, Z.; Aikawa, E.; Young-Pearse, T.; Kapogiannis, D. Detection of aggregation-competent tau in neuron-derived extracellular vesicles. Int. J. Mol. Sci. 2018, 19, 663.
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Carlson, O.D.; Mustapic, M.; Kapogiannis, D. Low neural exosomal levels of cellular survival factors in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 769–773.
- Sun, R.; Wang, H.; Shi, Y.; Gao, D.; Sun, Z.; Chen, Z.; Jiang, H.; Zhang, J. A Pilot Study of Urinary Exosomes in Alzheimer’s Disease. Neurodegener. Dis. 2019, 19, 184–191.
- Song, Z.; Xu, Y.; Zhang, L.; Zhou, L.; Zhang, Y.; Han, Y.; Li, X.; Yu, P.; Qu, Y.; Zhao, W. Comprehensive Proteomic Profiling of Urinary Exosomes and Identification of Potential Non-invasive Early Biomarkers of Alzheimer’s Disease in 5XFAD Mouse Model. Front. Genet. 2020.
- Rani, K.; Mukherjee, R.; Singh, E.; Kumar, S.; Sharma, V.; Vishwakarma, P.; Bharti, P.S.; Nikolajeff, F.; Dinda, A.K.; Goyal, V.; et al. Neuronal exosomes in saliva of Parkinson’s disease patients: A pilot study. Parkinsonism Relat. Disord. 2019, 67, 21–23.
- Guo, M.; Wang, J.; Zhao, Y.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Tieu, K. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain 2020, 143, 1476–1497.
- Yang, J.; Zhang, X.; Chen, X.; Wang, L.; Yang, G. Exosome mediated delivery of miR-124 promotes neurogenesis after ischemia. Mol. Ther. Nucleic Acids 2017, 7, 278–287.
- Lööv, C.; Scherzer, C.R.; Hyman, B.T.; Breakefield, X.O.; Ingelsson, M. α-Synuclein in extracellular vesicles: Functional implications and diagnostic opportunities. Cell. Mol. Neurobiol. 2016, 36, 437–448.
- Zhao, Z.-H.; Chen, Z.-T.; Zhou, R.-L.; Zhang, X.; Ye, Q.-Y.; Wang, Y.-Z. Increased DJ-1 and α-synuclein in plasma neural-derived exosomes as potential markers for Parkinson’s disease. Front. Aging Neurosci. 2019, 10, 438.
- Shi, M.; Liu, C.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C. Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol. 2014, 128, 639–650.
- Shi, M.; Kovac, A.; Korff, A.; Cook, T.J.; Ginghina, C.; Bullock, K.M.; Yang, L.; Stewart, T.; Zheng, D.; Aro, P. CNS tau efflux via exosomes is likely increased in Parkinson’s disease but not in Alzheimer’s disease. Alzheimer’s Dement. 2016, 12, 1125–1131.
- Zou, J.; Guo, Y.; Wei, L.; Yu, F.; Yu, B.; Xu, A. Long Noncoding RNA POU3F3 and α-Synuclein in Plasma L1CAM Exosomes Combined with β-Glucocerebrosidase Activity: Potential Predictors of Parkinson’s Disease. Neurother. J. Am. Soc. Exp. Neurother. 2020, 17, 1104–1119.
- Wang, S.; Liu, Z.; Ye, T.; Mabrouk, O.S.; Maltbie, T.; Aasly, J.; West, A.B. Elevated LRRK2 autophosphorylation in brain-derived and peripheral exosomes in LRRK2 mutation carriers. Acta Neuropathol. Commun. 2017, 5, 86.
- Fraser, K.B.; Moehle, M.S.; Daher, J.P.; Webber, P.J.; Williams, J.Y.; Stewart, C.A.; Yacoubian, T.A.; Cowell, R.M.; Dokland, T.; Ye, T. LRRK2 secretion in exosomes is regulated by 14-3-3. Hum. Mol. Genet. 2013, 22, 4988–5000.
- Reed, E.R.; Latourelle, J.C.; Bockholt, J.H.; Bregu, J.; Smock, J.; Paulsen, J.S.; Myers, R.H.; PREDICT-HD CSF Ancillary Study Investigators. MicroRNAs in CSF as prodromal biomarkers for Huntington disease in the PREDICT-HD study. Neurology 2018, 90, e264–e272.
- Denis, H.L.; Lamontagne-Proulx, J.; St-Amour, I.; Mason, S.L.; Weiss, A.; Chouinard, S.; Barker, R.A.; Boilard, E.; Cicchetti, F. Platelet-derived extracellular vesicles in Huntington’s disease. J. Neurol. 2018, 265, 2704–2712.
- Kumar, S.; Vijayan, M.; Bhatti, J.; Reddy, P.H. MicroRNAs as peripheral biomarkers in aging and age-related diseases. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2017; Volume 146, pp. 47–94.
- Gaughwin, P.M.; Ciesla, M.; Lahiri, N.; Tabrizi, S.J.; Brundin, P.; Björkqvist, M. Hsa-miR-34b is a plasma-stable microRNA that is elevated in pre-manifest Huntington’s disease. Hum. Mol. Genet. 2011, 20, 2225–2237.
- Xu, Q.; Zhao, Y.; Zhou, X.; Luan, J.; Cui, Y.; Han, J. Comparison of the extraction and determination of serum exosome and miRNA in serum and the detection of miR-27a-3p in serum exosome of ALS patients. Intractable Rare Dis. Res. 2018, 7, 13–18.
- Feneberg, E.; Steinacker, P.; Lehnert, S.; Schneider, A.; Walther, P.; Thal, D.R.; Linsenmeier, M.; Ludolph, A.C.; Otto, M. Limited role of free TDP-43 as a diagnostic tool in neurodegenerative diseases. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 351–356.
- Otake, K.; Kamiguchi, H.; Hirozane, Y. Identification of biomarkers for amyotrophic lateral sclerosis by comprehensive analysis of exosomal mRNAs in human cerebrospinal fluid. BMC Med. Genom. 2019, 12, 7.
- Banack, S.A.; Dunlop, R.A.; Cox, P.A. An miRNA fingerprint using neural-enriched extracellular vesicles from blood plasma: Towards a biomarker for amyotrophic lateral sclerosis/motor neuron disease. Open Biol. 2020, 10, 200116.
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005.
- Jeon, I.; Cicchetti, F.; Cisbani, G.; Lee, S.; Li, E.; Bae, J.; Lee, N.; Li, L.; Im, W.; Kim, M. Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol. 2016, 132, 577–592.
- Fisher, E.R.; Hayden, M.R. Multisource ascertainment of Huntington disease in Canada: Prevalence and population at risk. Mov. Disord. 2014, 29, 105–114.
- Morrison, P.; Harding-Lester, S.; Bradley, A. Uptake of Huntington disease predictive testing in a complete population. Clin. Genet. 2011, 80, 281–286.
- Zhang, X.; Abels, E.R.; Redzic, J.S.; Margulis, J.; Finkbeiner, S.; Breakefield, X.O. Potential transfer of polyglutamine and CAG-repeat RNA in extracellular vesicles in Huntington’s disease: Background and evaluation in cell culture. Cell. Mol. Neurobiol. 2016, 36, 459–470.
- Lee, M.; Liu, T.; Im, W.; Kim, M. Exosomes from adipose-derived stem cells ameliorate phenotype of Huntington’s disease in vitro model. Eur. J. Neurosci. 2016, 44, 2114–2119.
- Hong, Y.; Zhao, T.; Li, X.-J.; Li, S. Mutant huntingtin inhibits αB-crystallin expression and impairs exosome secretion from astrocytes. J. Neurosci. 2017, 37, 9550–9563.
- Corey-Bloom, J.; Haque, A.S.; Park, S.; Nathan, A.S.; Baker, R.W.; Thomas, E.A. Salivary levels of total huntingtin are elevated in Huntington’s disease patients. Sci. Rep. 2018, 8, 1–9.
- Coleman, B.M.; Hill, A.F. Extracellular vesicles–Their role in the packaging and spread of misfolded proteins associated with neurodegenerative diseases. Semin. Cell Dev. Biol. 2015, 40, 89–96.
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 1–19.
- Phukan, J.; Elamin, M.; Bede, P.; Jordan, N.; Gallagher, L.; Byrne, S.; Lynch, C.; Pender, N.; Hardiman, O. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: A population-based study. J. Neurol. Neurosurg. Psychiatry 2012, 83, 102–108.
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.-X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62.
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226.
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215.
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864.
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; Van Swieten, J.C.; Myllykangas, L. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268.
- Kim, H.J.; Kim, N.C.; Wang, Y.-D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A. Prion-like domain mutations in hnRNPs cause multisystem proteinopathy and ALS. Nature 2013, 495, 467.
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256.
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595.
- Aoki, Y.; Manzano, R.; Lee, Y.; Dafinca, R.; Aoki, M.; Douglas, A.G.L.; Varela, M.A.; Sathyaprakash, C.; Scaber, J.; Barbagallo, P.; et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 2017, 140, 887–897.
- Gomes, C.; Keller, S.; Altevogt, P.; Costa, J. Evidence for secretion of Cu, Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci. Lett. 2007, 428, 43–46.
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 2016, 139, 3187–3201.
- Maguire, G. Amyotrophic lateral sclerosis as a protein level, non-genomic disease: Therapy with S2RM exosome released molecules. World J. Stem Cells 2017, 9, 187.
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 2016, 139, 3187–3201.
- Maguire, G. Amyotrophic lateral sclerosis as a protein level, non-genomic disease: Therapy with S2RM exosome released molecules. World J. Stem Cells 2017, 9, 187.