The hypoxia signalling pathway enables adaptation of cells to decreased oxygen availability. When oxygen becomes limiting, the central transcription factors of the pathway, hypoxia-inducible factors (HIFs), are stabilised and activated to induce the expression of hypoxia-regulated genes, thereby maintaining cellular homeostasis.
- hypoxia
- HIF-1α
- HIF-2α
1. Introduction
Many pathways in mammalian cells rely on molecular oxygen; especially the final step of the mitochondrial respiratory chain. If oxygen levels drop below a cell-dependent critical level, cells experience hypoxia and cannot sustain aerobic respiration and subsequent ATP production. A switch to glycolysis, the less efficient but oxygen-independent pathway of producing ATP, is required to ensure cell survival. The switch between modes of energy generation and ultimately the adaption to a low oxygen environment, is controlled by the hypoxia signalling pathway. Hypoxia-inducible factors (HIFs), of which the first was described as a nuclear factor that enhances transcription of the erythropoietin (EPO) gene under hypoxic conditions by binding to a 3’ enhancer sequence element, are part of this pathway and maintain the adaptation at the transcriptional level [1][2]. HIFs consists of an oxygen-dependent α subunit that is destabilised in normoxia and a constitutively expressed β subunit (HIF-1β or ARNT) [3][4]. Three HIF-α subunits (HIF-1α, HIF-2α, HIF-3α) have been described, of which HIF-1α and HIF-2α are the best understood and considered as the major activators of hypoxia-induced gene transcription [2][5][6]. HIF-α and HIF-1β heterodimerise via their basic helix-loop-helix (bHLH)/Per-ARNT-Sim (PAS) domains to form the active transcription factor dimer, which then binds to hypoxia response elements (HREs) within the DNA of target genes [7]. HIFs then recruit general co-activators such as CBP/p300 via the C-terminal transactivation domain (C-TAD), leading to the expression of more than ~300 genes [8][9].
While HIF-1α and HIF-2α appear to be able to bind the same HRE, they can occupy distinct genomic sites, which vary with cell types. They display different subnuclear localisation and intranuclear diffusion speed [10]. In addition, some studies showed that neither HIF-1α nor HIF-2α could substitute the lack of DNA binding caused by the absence of the one or the other HIF-α variant [11]. However, others have shown that the loss of a single HIF-α isoform (either HIF-1α or HIF-2α) is compensated by the enhanced expression of the other and promote survival during cancer development [12]. This highlights the potential for specific contexts, whereby there may be a compensation mechanism when a single HIF-α isoform is silenced. These findings support, in part, the idea that HIF-1α accounts for acute and HIF-2α for chronic responses to hypoxia [13]. HIF-3α is less explored than HIF-1α or HIF-2α. HIF-3α mRNA is subject to alternative splicing in humans and in mice [14][15]. A specific mouse splice variant called inhibitory PAS domain protein (IPAS) was shown to interact directly with HIF-1α. The IPAS/HIF-1α complex was unable to bind to HREs and suggested to be a negative regulator of HIF-1α [16][17]. By contrast, the long human splice variant HIF-3α2 induces expression of various genes among them the EPO gene [18].
The canonical regulation of HIF-α protein stability and activity involves a series of molecular interactions and reversible covalent modifications to specific amino acids, termed post-translational modifications (PTMs) [19][20][21]. A PTM can alter a protein’s enzymatic activity, localisation, stability, and/or interaction with other proteins. Therefore, these non-genetically encoded modifications that are mainly carried out by enzymes, add to the complexity of the proteome as they can ascribe different functionalities to the same gene product. Common PTMs to a target protein include phosphorylation, acetylation, methylation, and alkylation as well as the covalent linkage of fatty acids, saccharides or small proteins such as ubiquitin and SUMO (small ubiquitin-related modifier) [22].
2. Canonical Regulation of HIF-α, the Role of Ubiquitination
Whilst regulation of HIF mRNA levels by hypoxia plays a minor role in HIF abundance, regulation of protein stability, via PTMs, is essential for appropriate HIF accumulation during hypoxia [23]. The HIF-α proteins are destabilised in normoxia via the ubiquitin-proteasome pathway (Figure 1) [24][25]. To achieve this, post-translational hydroxylation of two conserved proline residues (Pro-402/Pro-564 in HIF-1α; Pro-405/ Pro-531 in HIF-2α; P492 in HIF-3α) residing within an oxygen-dependent-degradation (ODD) domain is required [19][20][26][27]. Prolyl hydroxylation is carried out by three mammalian HIF prolyl hydroxylases (PHD-1, -2, and -3; also known as EglN2, EglN1, and EglN3, respectively). PHD2 acts as the main regulator of HIF-α degradation and has a key role in HIF-α intracellular dynamics [28][29]. Once hydroxylated, the HIF-α proteins are recognised by an E3-ligase complex containing the von-Hippel-Lindau protein (pVHL), which acts as the substrate recognition unit and, together with Cullin-2 (Cul-2), Elongin-1, Elongin-2 and Ring-Box 1 (RBX1), polyubiquitinates K532, K538, or K567 on HIF-1α (and K497, K503, or K512 on HIF-2α) [30][31][32][33]. The polyubiquitylated HIF-α proteins are then degraded via the 26S proteasome [19]. When cells are deprived of oxygen, PHDs have less molecular oxygen available to act as a co-factor, hence decreasing their activity and subsequent HIF-α hydroxylation [34]. Consequently, HIF-α subunits accumulate in the nucleus, form a heterodimer with the constitutively expressed HIF-1β and bind to HREs [7]. HIF-α hydroxylation is not exclusively mediated via PHDs, but also by the Factor-Inhibiting HIF (FIH). FIH is an asparaginyl hydroxylase and modifies HIF-1α and HIF-2α on their C-TAD residues N803 and N847, respectively [35][36]. During hypoxia, FIH activity is suppressed, allowing HIF-1α or -2α to complex with the CBP/p300 co-activators and increased transcriptional activation. HIF-α hydroxylation has long been considered to be an irreversible PTM, but it was recently shown, by mass spectrometry, that the FIH-mediated asparagine hydroxylation is indeed a reversible process [37].
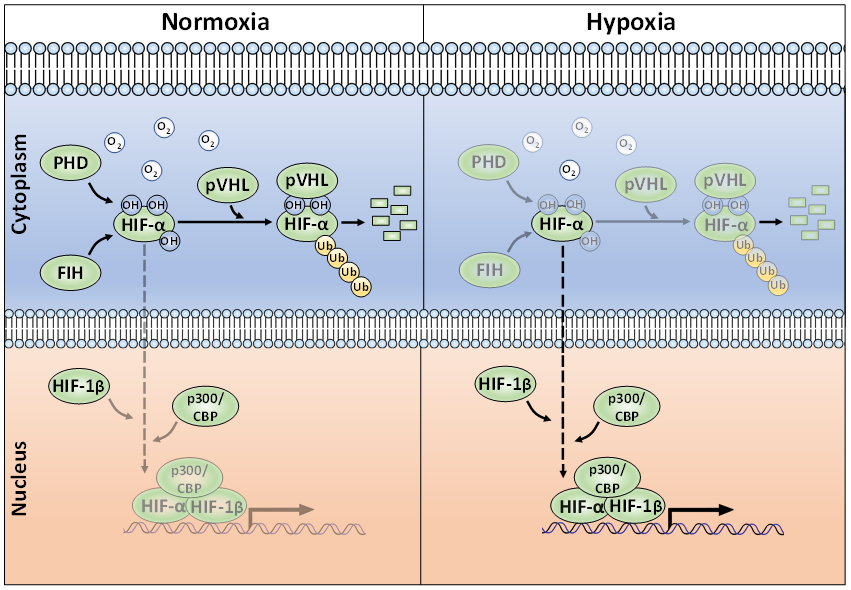
Figure 1. The canonical oxygen-dependent degradation mechanism for HIF-α by pVHL-mediated 26S proteasomal degradation and its inhibition by Factor-Inhibiting HIF (FIH) during hypoxia.
Polyubiquitination is a quintessential PTM in preventing unwanted HIF-α accumulation in normoxia. Aside from ubiquitination, the action of deubiquitinating enzymes (termed DUBs) are well-known for their roles in regulating HIF-α. The action of DUBs on HIFs has been extensively and recently reviewed and will not be covered here [38][39].
3. Non-Canonical PTMs Regulating HIF-α Subunits
Whilst there are many well-characterised binding partners and indirect regulators of HIF-1α and HIF-2α, PTMs constitute an essential direct regulatory mechanism for the HIF transcription factors [40]. PTMs abundantly decorate the full-length of these oxygen-sensitive proteins to exert specific regulatory forces. Most of these covalent modifications are enzymatically driven, with some exceptions such as S-nitrosylation. For the past two decades, the PTM landscape of HIF-1α and HIF-2α has been ever-expanding, showing the intrinsic complexity and crosstalk of diverse intracellular signalling pathways implicating HIF activity, stability, and localisation (Figure 2).
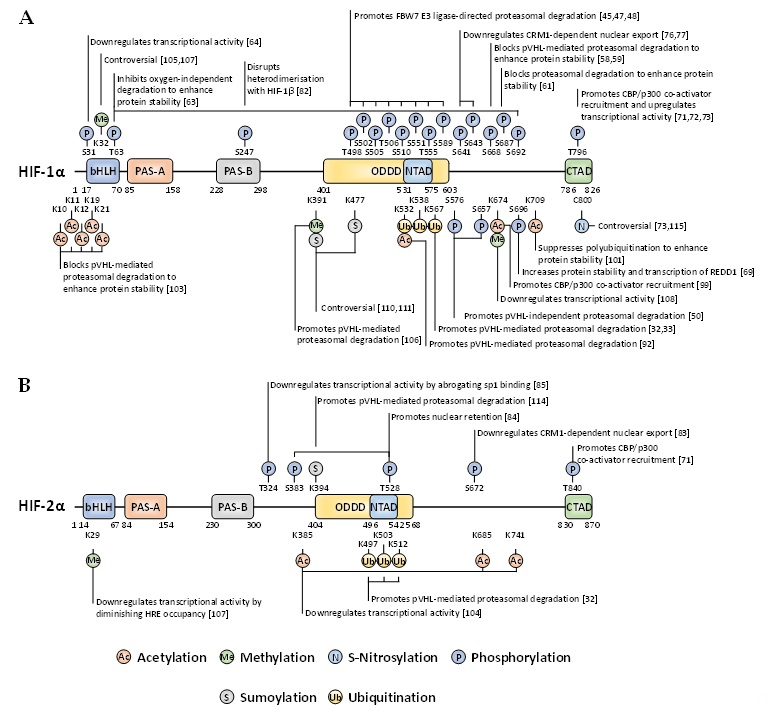
Figure 2. Localisation and function of non-canonical HIF-α post-translational modifications (PTMs) mapped onto full-length HIF-1α (A) and HIF-2α (B). When a given PTM has multiple publications stating conflicting functional outcomes, then the functionality is denoted as ‘controversial’.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22010268
References
- Semenza, G.L.; Nejfelt, M.K.; Chi, S.M.; Antonarakis, S.E. Hypoxia-inducible nuclear factors bind to an enhancer element
- located 3' to the human erythropoietin gene. Natl. Acad. Sci. USA 1991, 88, 5680–5684, doi:10.1073/pnas.88.13.5680.
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human
- erythropoietin gene enhancer at a site required for transcriptional activation. Cell. Biol. 1992, 12, 5447–5454, doi:10.1128/mcb.12.12.5447.
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer
- regulated by cellular O2 tension. Natl. Acad. Sci. USA 1995, 92, 5510–5514, doi:10.1073/pnas.92.12.5510.
- Wang, G.L.; Semenza, G.L. Purification and characterization of hypoxia-inducible factor 1. Biol. Chem. 1995, 270, 1230–1237, doi:10.1074/jbc.270.3.1230.
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Natl. Acad. Sci. USA 1997, 94, 4273–4278, doi:10.1073/pnas.94.9.4273.
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 1998, 7, 205–213.
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. STKE 2005, 2005, re12, doi:10.1126/stke.3062005re12.
- Arany, Z.; Huang, L.E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Natl. Acad. Sci. USA 1996, 93, 12969–12973, doi:10.1073/pnas.93.23.12969.
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669, doi:10.1182/blood-2004-07-2958.
- Taylor, S.E.; Bagnall, J.; Mason, D.; Levy, R.; Fernig, D.G.; See, V. Differential sub-nuclear distribution of hypoxia-inducible factors (HIF)-1 and -2 alpha impacts on their stability and mobility. Open Biol. 2016, 6, doi:10.1098/rsob.160195.
- Smythies, J.A.; Sun, M.; Masson, N.; Salama, R.; Simpson, P.D.; Murray, E.; Neumann, V.; Cockman, M.E.; Choudhry, H.; Ratcliffe, P.J.; et al. Inherent DNA-binding specificities of the HIF-1alpha and HIF-2alpha transcription factors in chromatin. EMBO Rep. 2019, 20, doi:10.15252/embr.201846401.
- Menrad, H.; Werno, C.; Schmid, T.; Copanaki, E.; Deller, T.; Dehne, N.; Brune, B. Roles of hypoxia-inducible factor-1alpha (HIF-1alpha) versus HIF-2alpha in the survival of hepatocellular tumor spheroids. Hepatology 2010, 51, 2183–2192, doi:10.1002/hep.23597.
- Lofstedt, T.; Fredlund, E.; Holmquist-Mengelbier, L.; Pietras, A.; Ovenberger, M.; Poellinger, L.; Pahlman, S. Hypoxia inducible factor-2alpha in cancer. Cell Cycle 2007, 6, 919–926, doi:10.4161/cc.6.8.4133.
- Makino, Y.; Kanopka, A.; Wilson, W.J.; Tanaka, H.; Poellinger, L. Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3 alpha locus. Biol. Chem. 2002, 277, 32405–32408, doi:10.1074/jbc.C200328200.
- Pasanen, A.; Heikkila, M.; Rautavuoma, K.; Hirsila, M.; Kivirikko, K.I.; Myllyharju, J. Hypoxia-inducible factor (HIF)-3a is subject to extensive alternative splicing in human tissues and cancer cells and is regulated by HIF-1 but not HIF-2. J.
- Biochem. Cell Biol. 2010, 42, 1189–1200, doi:10.1016/j.biocel.2010.04.008.
- Heikkila, M.; Pasanen, A.; Kivirikko, K.I.; Myllyharju, J. Roles of the human hypoxia-inducible factor (HIF)-3 alpha variants in the hypoxia response. Mol. Life Sci. 2011, 68, 3885–3901, doi:10.1007/s00018-011-0679-5.
- Makino, Y.; Uenishi, R.; Okamoto, K.; Isoe, T.; Hosono, O.; Tanaka, H.; Kanopka, A.; Poellinger, L.; Haneda, M.; Morimoto, C. Transcriptional up-regulation of inhibitory PAS domain protein gene expression by hypoxia-inducible factor 1 (HIF-1)—A negative feedback regulatory circuit in HIF-1-mediated signaling in hypoxic cells. Biol. Chem. 2007, 282, 14073–14082, doi:10.1074/jbc.M700732200.
- Tolonen, J.P.; Heikkila, M.; Malinen, M.; Lee, H.M.; Palvimo, J.J.; Wei, G.H.; Myllyharju, J. A long hypoxia-inducible factor 3 isoform 2 is a transcription activator that regulates erythropoietin. Mol. Life Sci. 2020, 77, 3627–3642, doi:10.1007/s00018-019-03387-9.
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468, doi:10.1126/science.1059817.
- Jaakkola, P.; Mole, D.R.; Tian, Y.M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472, doi:10.1126/science.1059796.
- Masson, N.; Willam, C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001, 20, 5197–5206, doi:10.1093/emboj/20.18.5197.
- Ribet, D.; Cossart, P. Ubiquitin, SUMO, and NEDD8: Key Targets of Bacterial Pathogens. Trends Cell. Biol. 2018, 28, 926–940, doi:10.1016/j.tcb.2018.07.005.
- Gorlach, A. Regulation of HIF-1alpha at the transcriptional level. Pharm. Des. 2009, 15, 3844–3852, doi:10.2174/138161209789649420.
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. Biol. Chem. 1996, 271, 32253–32259, doi:10.1074/jbc.271.50.32253.
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. Biol. Chem. 1997, 272, 22642–22647, doi:10.1074/jbc.272.36.22642.
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Natl. Acad. Sci. USA 1998, 95, 7987–7992, doi:10.1073/pnas.95.14.7987.
- Maynard, M.A.; Qi, H.; Chung, J.; Lee, E.H.L.; Kondo, Y.; Hara, S.; Conaway, R.C.; Conaway, J.W.; Ohh, M. Multiple splice variants of the human HIF-3 alpha locus are targets of the von Hippel-Lindau E3 uhiquitin ligase complex. Biol. Chem. 2003, 278, 11032–11040, doi:10.1074/jbc.M208681200.
- Berra, E.; Benizri, E.; Ginouves, A.; Volmat, V.; Roux, D.; Pouyssegur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1 alpha in normoxia. EMBO J. 2003, 22, 4082–4090, doi:10.1093/emboj/cdg392.
- Bagnall, J.; Leedale, J.; Taylor, S.E.; Spiller, D.G.; White, M.R.; Sharkey, K.J.; Bearon, R.N.; See, V. Tight control of
- hypoxia-inducible factor-alpha transient dynamics is essential for cell survival in hypoxia. Biol. Chem. 2014, 289, 5549–5564, doi:10.1074/jbc.M113.500405.
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275, doi:10.1038/20459.
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.Y.; Huang, L.E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Cell. Biol. 2000, 2, 423–427, doi:10.1038/35017054.
- Paltoglou, S.; Roberts, B.J. HIF-1alpha and EPAS ubiquitination mediated by the VHL tumour suppressor involves flexibility in the ubiquitination mechanism, similar to other RING E3 ligases. Oncogene 2007, 26, 604–609, doi:10.1038/sj.onc.1209818.
- Tanimoto, K.; Makino, Y.; Pereira, T.; Poellinger, L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000, 19, 4298–4309, doi:10.1093/emboj/19.16.4298.
- Stiehl, D.P.; Wirthner, R.; Koditz, J.; Spielmann, P.; Camenisch, G.; Wenger, R.H. Increased prolyl 4-hydroxylase domain
- proteins compensate for decreased oxygen levels. Evidence for an autoregulatory oxygen-sensing system. Biol. Chem. 2006, 281, 23482–23491, doi:10.1074/jbc.M601719200.