DNA glycosylases are a set of enzymes responsible for initiating the base excision repair pathway thereby contributing to the maintenance of the genome. They are responsible for the eradication of the vast number of small, non-helical-distorting base lesions from the genome, resulting from oxidative, alkylating, and deamination events, induced by environmental factors such as ionizing radiation, endogenous factors such as reactive oxygen species (ROS), or anticancer agents such chemotherapeutic drugs or radiotherapy. Because of their key role in DNA repair, they are now considered as potential anti-cancer drug targets and efforts are being made worldwide to identify potent inhibitors of these enzymes that could be used to potentiate classical chemo- or radio-therapy.
- DNA repair
- Enzyme
- Base excision repair
- Anti-cancer drug target
1. DNA glycosylases and Base excision repair
Cells have evolved five major elaborate DNA damage repair pathways to detect and repair the wide diversity of DNA lesions, namely base excision repair (BER), nucleotide excision repair (NER), homologous recombination (HR), non-homologous end-joining (NHEJ), and mismatch repair. Each pathway is responsible for the repair of a different type of DNA damage and together they maintain genomic integrity and stability [1][2][3]. BER is an essential DNA repair pathway that contributes to the stability of the genome by eradicating the vast number of small, non-helical-distorting base lesions from the genome, resulting from oxidative, alkylating, and deamination events, induced by environmental factors, endogenous factors such as reactive oxygen species (ROS), or anticancer agents such chemotherapeutic drugs or radiotherapy [4][5]. Among the damaged bases repaired by the BER pathway (recently reviewed in [6]), one can cite the highly produced and mutagenic oxidized base 8-oxo-guanine (8-oxo-G), the lethal thymine glycols (Tg) caused by oxidation of thymine bases, which induce replication and transcription blockade, uracil misincorporation in DNA, abasic sites, and single-strand breaks (SSB) [7][8].
The BER pathway processes damaged bases in a series of successive reactions to eliminate all the intermediate products that otherwise block replication (Figure 1). The BER pathway is initiated by a set of enzymes, known as DNA glycosylases, responsible for identifying and eliminating the damaged bases, thereby generating an apurinic/apyrimidinic (AP) site [9][10][11]. DNA glycosylases bind to damaged bases and induce the aberrant base to flip out of the double helix and enter the binding site of the enzyme [10][12][13]. They then catalyze the cleavage of the N-glycosidic bond between the substrate base and the 2′-deoxyribose to efficiently remove the damaged base [11][14]. DNA glycosylases can be subdivided into either monofunctional or bifunctional enzymes depending on their catalytic activities (Figure 1). Monofunctional DNA glycosylases like uracil-N glycosylase (UNG) exhibit only DNA glycosylase activity and produce an AP-site, which is further processed by an AP endonuclease, APE1, in humans. In contrast, bifunctional glycosylases, like endonuclease III-like 1 (NTH1) and endonuclease VIII-like 1-3 (NEIL1-3) glycosylases, exhibit both DNA glycosylase and AP-lyase activities. After release of the damaged base, bifunctional DNA glycosylases with a β-lyase activity form a transient Schiff base intermediate between the DNA and an active site lysine residue, after which the sugar-phosphate backbone is nicked via a β-elimination reaction to create a 3′ α,β-unsaturated aldehyde and a 5′ phosphate group [15][16] that are further processed by APE1 and a polynucleotide kinase/phosphatase (PNKP), respectively. An amino-terminal proline residue provides NEIL1 and NEIL2 with an additional β-δ-elimination activity and makes APE1, but not PNKP, dispensable in these cases [17][18].
The subsequent BER enzymes then repair the AP-containing DNA damage [4]. An AP endonuclease (APE1) or an AP lyase cleaves the DNA backbone and produces either a single-stranded DNA (ssDNA) nick 5′ to the AP site in the case of AP endonucleases, or 3′ to the AP site for AP lyases (Figure 1). The DNA ends are then processed by one of two sub-pathways, short- or long-patch repair, during which the DNA polymerase fills the gap with the correct nucleotides and the repair mechanism is completed after sealing of the nick by a DNA ligase [14]. These end processing steps take place by different mechanisms depending on the type of DNA glycosylase, the physiological state of the cell, and the availability of BER factors [4]. In short-patch BER, which is used in proliferating and non-proliferating cells, an AP endonuclease removes the 3′-deoxyribose phosphate (dRP) after strand cleavage and PNKP removes the 3′-phosphate, and the single nucleotide gap is then filled and ligated by DNA polymerase β and DNA ligase I or III [4][5]. In addition, PARP1 and XRCC1 cofactors participate in some types of short-patch repair. Long-patch BER, which takes place mainly in proliferating cells, makes use of several additional replication proteins, to fill 2–10 nucleotide gaps. These include DNA polymerase δ/ε, PCNA, the flap endonuclease FEN1, and DNA ligase I [4][5].
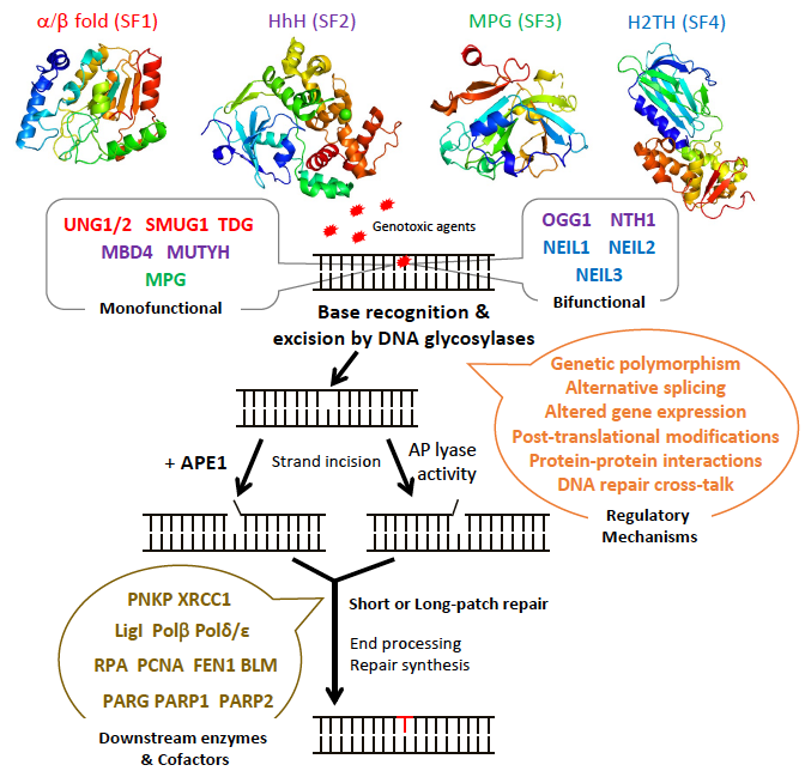
Figure 1. Schematic diagram illustrating the steps and enzymes involved in the base excision repair (BER) pathway. Representative structures of the different superfamilies (SF) of DNA glycosylases (SF1, α/β fold family, red; SF2, helix-hairpin-helix (HhH) family, purple; SF3, 3-methyl-purine glycosylase (MPG) family, green; SF4, helix-two-turn-helix (H2TH) family, blue) responsible for recognition and removal of damaged bases are shown. After cleavage of the damaged strand by an apurinic/apyrimidinic (AP) endonuclease, APE1, or by the AP lyase activity of bifunctional DNA glycosylases, downstream BER enzymes together with several cofactors (listed in brown) prepare the damaged site for de novo synthesis using one of two sub-pathways: short-patch or long-patch repair. DNA glycosylases are tightly regulated at the gene, mRNA, and protein levels by a set of regulatory systems (listed in orange). UNG1/2: uracil-N glycosylase 1 or 2; SMUG1: single-strand-specific monofunctional uracil DNA glycosylase 1; TDG: thymine DNA glycosylase; MBD4: methyl-CpG-binding protein 4; MUTYH: MutY homolog DNA glycosylase; OGG1: 8-oxo-G DNA glycosylase 1; NTH1: endonuclease III-like 1; NEIL1-3: endonuclease VIII-like 1-3.
DNA glycosylases have been classified into four superfamilies (SF) that reflect their functional activities and their structural features (Figure 1) [4][19][20]. SF1 comprises uracil DNA glycosylases (UDG), such as UNG, single-strand-specific monofunctional uracil DNA glycosylase 1 (SMUG), and thymine DNA glycosylase (TDG). These glycosylases possess a characteristic α/β fold and target the removal of uracil (U) formed by the deamination of cytosine caused by oxidative stress. UNG and SMUG1 have similar substrate specificity as they both remove mis-incorporated U in DNA. UNG1 and UNG2 are two splice variants of UNG, which localize respectively to mitochondria and the nucleus [21]. Beyond their role in the error-free repair of U in DNA, UNG2, and also as shown more recently, UNG1, play a key, mutagenic role in somatic hypermutation and class switch recombination during B cell receptor/antibody maturation, two processes that involve deamination of cytidines to uridines in ssDNA [22][23][24]. TDG is capable of removing oxidized or deaminated pyrimidine bases. It has also been reported to participate in the epigenetic demethylation signaling pathway as part of the multistep pathway that removes the methyl group of cytosine within CpG sites [25][26]. The second superfamily (SF2) comprises DNA glycosylases with a characteristic helix-hairpin-helix (HhH) motif, which include glycosylases such as NTH1, 8-oxo-G DNA glycosylase 1 (OGG1), MutY homolog DNA glycosylase (MUTYH), and methyl-CpG-binding protein 4 (MBD4). This family of glycosylases targets various lesions caused by oxidative stress including 8-oxo-G, Tg, 4,6-diamino-5-formamidopyrimidine (FapyA), and 2,6-diamino-4-hydroxy-5-formamido-pyrimidine (FapyG). SF3 comprises only one member, which is the 3-methyladenine-DNA glycosylase also called 3-methyl-purine glycosylase (MPG). Unlike the other superfamilies, MPG is not characterized by a specific fold and targets the damage caused by alkylation instead of oxidative stress. In addition to excising 3-methyladenine (3MeA), MPG also excises DNA bases with methyl or certain other alkyl groups at the N7 and N3 positions of both adenine and guanine [27]. Finally, the fourth superfamily (SF4) comprises NEIL glycosylases, including NEIL1, NEIL2, and NEIL3. The characteristic fold of this superfamily is a helix-two-turn-helix (H2TH) motif. This superfamily is similar to HhH SF2 in function as it targets the oxidative base damages such as FapyG and FapyA [28]. Within each of these four superfamilies, each enzyme displays a distinct, but partially overlapping substrate specificity. This partially explains why most DNA glycosylase knockout mice are viable and do not present clear phenotypes, except for TDG null mice that show an embryonic lethal phenotype, most likely as a result of its impaired epigenetic function [20][29]. Some DNA glycosylases like UNG, SMUG1, MPG, and NEIL1 can recognize damaged bases both in double-stranded (dsDNA) and in ssDNA, whereas DNA glycosylases like TDG, MBD4, OGG1, and MUTYH only recognize base lesions in dsDNA.
2. DNA Glycosylases as anti-cancer drug targets
With the exception of surgery, all major anticancer therapeutic strategies, including radio-, chemo-, and immuno-therapies, used alone or in combination, aim to specifically kill cancer cells within affected tissues [30][31][32]. Cytotoxic chemotherapy, developed 60 years ago, still represents a widely used treatment and in many cases involves the administration of powerful genotoxic agents acting either directly (e.g., platinum-based drugs) or indirectly (e.g., topoisomerase inhibitors) on DNA in cells that rapidly proliferate [33][34][35]. There are several ways in which base lesions are generated in cancer cells. Base lesions can be intrinsically acquired due to altered metabolic pathways that produce an excess of ROS, leading to oxidation of DNA bases. Base lesions can also be caused directly by chemotherapeutic agents reacting with DNA as in the case of alkylating agents, and can be generated indirectly by oxidative stress provoked by chemotherapeutic drugs such as cisplatin. By repairing all of these DNA lesions, the BER pathway reduces the cytotoxic effects of anticancer drugs, thereby contributing to the survival of cancer cells [4][6]. As a result, BER enzymes are increasingly considered as valid targets for cancer treatment [30][7][36][37]. However, because of the partial redundancy of DNA glycosylases and the cross-talk between different repair pathways, the efficiency of DNA glycosylase inhibitors as monotherapy is expected to be limited, but should be greatly enhanced when used either in combination therapy together with a conventional DNA-damaging chemotherapeutic agent, or in a more personalized medicine approach, for example in tumors displaying defects in alternative repair pathways (synthetic lethality concept). Currently, most efforts have aimed to identify small molecule inhibitors of the catalytic activities of DNA glycosylases. Different approaches have been used with success: (i) Targeted low-throughput approaches, (ii) computational- and structure-based rational drug design, and (iii) high-throughput screening (HTS) of chemical or fragment-based libraries, all of which have recently been shown to be valid and complementary strategies to find potent inhibitors of DNA glycosylases [38].
Several metabolic cofactors have been shown to efficiently inhibit DNA glycosylases. This is the case of pyridoxal 5′-phosphate (PLP), a cofactor of enzymes involved in amino acid metabolism. The aldehyde moiety of PLP has been shown to inhibit or inactivate diverse DNA-dependent enzymes and was therefore tested on several DNA glycosylases. Of these, only NEIL2 was significantly inhibited by PLP due to the formation of a Schiff base between PLP and a DNA-binding loop in the enzyme [39]. Similarly, the intermediate of tyrosine catabolism, fumarylacetoacetate (FAA), was also found to specifically inhibit a subset of DNA glycosylases. In particular, the NEIL1 and NEIL2 enzymes were strongly inhibited, whereas only a small effect was observed in vitro on NTH1 and OGG1 and no effect on UNG2 [40]. FAA inhibition of DNA glycosylases may explain the increased mutagenesis rates associated with hepatocarcinoma development in HT1 patients, which have a deficiency in FAA hydrolase and thus accumulate high intracellular levels of this intermediate catabolite [40].
Given the availability of high-resolution crystallographic structures of several human DNA glycosylases (UNG, TDG, OGG1, NEIL1, NEIL3, MUTYH, MPG, and MBD4), computational modeling and structure-based drug design have also been used to identify and optimize DNA glycosylase inhibitors. A recent computational druggability assessment study revealed that DNA glycosylases are druggable targets, with OGG1, MUTYH, NEIL1, UNG, and TDG being the most favorable drug-binding proteins [38]. Structural studies of bacterial formamidopyrimidine-DNA glycosylase (Fpg) and human NEIL enzymes are currently been exploited, for example, to develop NEIL1 inhibitors derived from the 2-thioxanthine (2TX) compound that was originally found to specifically inhibit bacterial Fpg and not its human homolog [41][42]. Structure-based protein engineering has been used to improve the selectivity of SAUGI, an inhibitory protein from Staphylococcus aureus, for human UDG versus Herpes simplex virus (HSV) UDG by comparing the crystal structures of SAUGI-human UDG with that of SAUGI-HSV-UDG [43]. Structural studies have also guided the optimization of fragment-based inhibitors of human UDG [44].
To identify UNG inhibitors, Jiang and colleagues developed a uracil-directed ligand tethering strategy, in which a uracil-aldehyde ligand was tethered via alkyloxyamine linker chemistry to a diverse array of aldehyde binding elements. The goal was to exploit the uracil ligand to target the UNG active site and the alkyloxyamine linker tethering to randomly explore peripheral binding pockets. This original approach rapidly identified the first small molecule inhibitors of human UNG with micromolar to submicromolar binding affinities [45] and the best inhibitor was co-crystallized with the UNG2 enzyme. The structure of inhibitor-bound UNG2 revealed that the inhibitor engages in crucial electrostatic interactions and hydrogen bonding with the enzyme, similar to those seen when complexed to uracil-containing DNA [46].
Over the past decade, novel fluorescence-based HTS approaches have been developed to identify DNA glycosylase inhibitors. These assays respond to three essential criteria: Fast, robust, and adaptable (Figure 2). The use of such approaches has led to the selection and validation of several efficient drugs against DNA glycosylases. In a recent and precursor study, Jacobs et al. developed a fluorescence-based assay adapted for HTS to find inhibitors of NEIL1 [47], which has been identified as a possible candidate for a synthetic lethality approach in Fanconi anemia disease [48]. This first study was inspired by an HTS for inhibitors of APE1 [49]. It is designed to select inhibitors of bifunctional DNA glycosylases, as it is based on strand incision activity that occurs after hydrolysis of the glycosylic bond. In this assay, a short synthetic oligodeoxyribonucleotide containing secondary oxidation products of 8-oxo-G (spirodihydantoin, Sp, and guanidinohydantoin, Gh) and labeled at its 5′ end with the TAMRA red fluorophore was hybridized to a complementary DNA containing a quencher molecule at its 3′ end (Figure 2A). Upon repair of the site-specific lesion by NEIL1, the DNA fragment containing the TAMRA is incised and released from the DNA duplex, leading to increased fluorescence. The relative fluorescence can then be measured both in the presence and absence of the inhibitor. HTS led to the selection of purine analogs with IC50 values ranging from 4 to 25 μM. Interestingly, four of them significantly inhibited the in vitro repair activity of NEIL1 on γ-irradiated calf thymus DNA. Although the selected inhibitors were also shown to significantly block the activity of closely-related DNA glycosylases such as NTH1, this study established the first customized fluorescence-based assay for HTS to find inhibitors of DNA glycosylases.
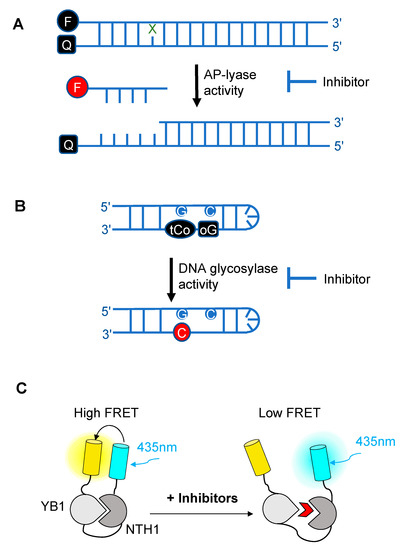
Figure 2. Schematic diagrams illustrating the different fluorescence-based assays developed over the past decade for high-throughput screening (HTS) of chemical libraries for the selection of inhibitors of either the AP-lyase activity [50] (A), the DNA glycosylase activity [51] (B) of DNA glycosylases, or of the interaction interface (C) between a DNA glycosylase (here, NTH1) and its cellular partner (YB1) [52]. FRET: Förster resonance energy transfer. In (A), X denotes a damaged base processed by DNA glycosylases and Q denotes the quencher of the fluorophore (black F). Cleavage by the AP lyase activity results in the release of the fluorophore-labeled lesion-containing strand and fluorescence emission (red F). In (B), release of a modified 8-oxo-G base (oG) linked to a quencher that specifically quenches the highly fluorescent DNA base analogue, tCo, covalently bound to the neighboring base, leads to fluorescence emission (red C). In (C), NTH1-YB1 complex formation is associated with high FRET levels, which are significantly reduced by inhibitors (red wedge) of the PPI interface.
As OGG1 is an essential bifunctional DNA glycosylase responsible for removing the most abundant oxidized base produced in cells, it is now considered as a candidate of choice for the development of anticancer drugs. To do this, the fluorescence-based assay developed by Jacobs and colleagues was adapted to OGG1 [53]. The authors identified hydrazide or acyl hydrazine-based inhibitors that display submicromolar IC50 values against OGG1 incision strand activity. These very encouraging results were further confirmed by conventional gel shift assays, confirming the potential of such HTS using miniaturized assays to find small molecule inhibitors of DNA glycosylases. Moreover, the selected inhibitors were found to be specific to OGG1; little to no inhibition of NEIL1, NTH1, and bacterial Fpg assessed on their respective substrates (FapyG for NEIL1 and NTH1 and 8-oxo-G for Fpg) was detected.
The assays described above, however, are not suitable for measuring the monofunctional glycosylase activity of DNA glycosylases since they rely on the release of the fluorophore-labeled strand after strand incision by the AP lyase activity. In vivo, OGG1, like NTH1, has been shown to act predominantly as a monofunctional DNA glycosylase [54]; thus, the assay presented in Figure 2A may not be the most adapted to find potent OGG1 inhibitors. Different technical solutions have been found to select inhibitors of the DNA glycosylase activity. Mancuso and colleagues added APE1 to their molecular beacon-based assay to detect the monofunctional glycosylase activity of TDG [55]. They screened 2000 drugs and identified 20 candidate inhibitors. Some of these compounds, like juglone and closantel, were confirmed as TDG inhibitors in a standard DNA glycosylase assay. These compounds also led to a dose-dependent reduction in cell viability of melanoma cells with IC50 values close to 10 µM, which suggests that interfering with the DNA repair and epigenetic activity of TDG may represent a new and valid approach for the treatment of melanoma. A similar strategy was also used recently in a search for new therapeutic strategies to fight inflammation. Visnes and colleagues selected a site-specific inhibitor of OGG1, named TH5487, which blocks OGG1’s ability to recognize and repair 8-oxo-G containing DNA [56]. A recent crystal structure of human OGG1 bound to this inhibitor (unpublished; PDB 6RLW) reveals the intricate interactions formed between the inhibitor and the substrate binding pocket of OGG1 (Figure 3A,B).
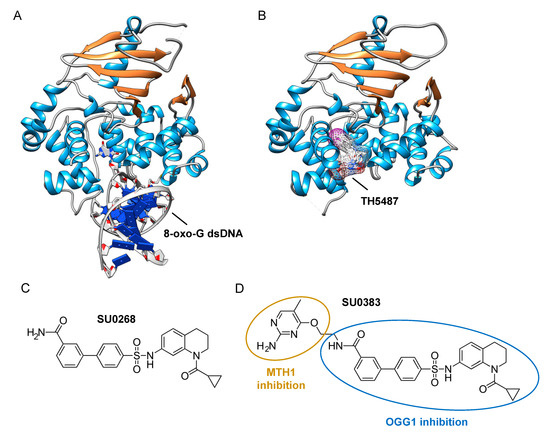
Figure 3. Identification of specific inhibitors of human OGG1. (A) Crystal structure of human OGG1 crosslinked to 8-oxo-G containing dsDNA (PDB: 6W0M; [57]). (B) Crystal structure of human OGG1 bound to an active site-specific inhibitor, TH5487 (PDB 6RLW), represented in ball and sticks and as a transparent mesh. (C) Chemical structure of the highly specific OGG1 inhibitor selected by Tahara and colleagues [51], SU0268. (D) Chemical structure of the dual inhibitor, SU0383, which efficiently blocks both MTH1 and OGG1 activities [58].
Tahara and colleagues used a different strategy to target the glycosylase activity of OGG1. They adapted a previously reported fluorogenic assay [59] to study the release of 8-oxo-G from DNA [51]. This assay makes use of a fluorescent hairpin oligomer probe, called OGR1 [60], containing a modified 8-oxo-G base linked to a quencher that specifically quenches a highly fluorescent DNA base analogue, tCo, covalently bound to the base of the neighboring nucleotide (Figure 2B). After cleavage and release of the 8-oxo-G moiety and its associated quencher, the tCo molecule on the oligonucleotide becomes fluorescent and the fluorescent signal can be followed in real time. In this assay, only the DNA glycosylase activity is measured, allowing us to specifically screen and select drugs that interfere with the break-down of the N-glycosidic link between the 8-oxo-G base and the deoxyribose. After HTS, one compound named SU0268, which has an acyl tetrahydroquinoline sulfonamide skeleton with an IC50 of 59 nM was selected (Figure 3C). This competitive inhibitor is highly specific to OGG1, shows no cytotoxicity in cells, and inhibits OGG1 activity both in vitro and in cells, where the abundance of 8-oxo-G bases in cells incubated with 0.5 µM SU0268 increased to levels comparable to those detected in cells treated with 0.5 H2O2 and 0.3 mM of Cr3+ [51].
Upon oxidative stress, dGTP is in part transformed into 8-oxo-dGTP that can be incorporated into DNA. To prevent this, a nudix hydrolase, the MutT homolog-1 (MTH1) enzyme converts 8-oxo-dGTP into 8-oxo-dGMP, which can no longer be incorporated into DNA [61]. Recently, MTH1 was also identified as an interesting target for inhibitor screening in the context of cancer, and several small molecule drugs were identified [62]. Based on these studies, Tahara and colleagues set out to design a dual inhibitor corresponding to the association of two molecules, one specific for MTH1 and the other being SU0268 (the inhibitor of OGG1), giving rise to compound SU0383 (Figure 3D). This small molecule inhibits OGG1 with an IC50 of 49 nM, measured using the OGR1 probe. It also inhibits MTH1 with an IC50 of 34 nM, using a luminescence-based assay for the activity of MTH1 in the conversion of 8-oxo-dGTP to 8-oxo-dGMP [63]. Interestingly, exposure of MCF7 breast cancer cells to 16 µM of H2O2 in the presence of SU0383 leads to a drop in cell viability of 20%, demonstrating the ability of the molecule to increase the sensitivity of the cells towards oxidative stress [58].
In a recent study conducted by Senarisoy et al., an alternative approach was proposed to restore sensitivity of drug-resistant tumor cells, by targeting the interaction of NTH1 with one of its cellular partners, YB1, instead of targeting the catalytic activities of NTH1 [52]. YB1 is a transcription factor that has been shown to bind to NTH1 and stimulate its AP-lyase activity. In drug-resistant tumor cells, nuclear localization of YB1 is elevated and this favors the interaction of NTH1 with YB1, leading to an increased abundance of the NTH1-YB1 complex. In this study, inhibitors of the NTH1-YB1 interaction were identified and validated using a FRET-based biosensor that was designed for HTS (Figure 2C). In this biosensor construct, the fluorescent protein, sYFP2, was placed at the amino terminus followed by YB1 and NTH1, and a second FRET-compatible fluorescent protein, mTQ2, at the C-terminus. Using this construct, HTS of 1200 molecules was performed and 8 potent inhibitor molecules were selected. The inhibitory effects of these molecules were further confirmed by AlphaLISA and their molecular targets were identified using thermal shift assay. Of the 8 inhibitors, two molecules, meclocycline and oxytetracycline, were found to have low IC50 values, i.e., 1.5 and 10.3 μM. To further investigate their inhibitory properties in drug-resistant tumor cells, cisplatin-resistant MCF7 cells were used and the effects of the inhibitor molecules on the sensitivity of MCF7 cells to cisplatin was studied. The results of this study show that both meclocycline and oxytetracycline induced a small, but significant, concentration-dependent decrease in the viability of MCF7 cells treated with cisplatin, indicating that these molecules partially restore the sensitivity of resistant MCF7 cells to cisplatin [52]. This study thus demonstrates that the PPIs involving DNA glycosylases also constitute druggable targets for the development of new therapeutic strategies to eliminate cancer cells.
3. Conclusion
It is now clear that DNA glycosylases are key enzymes that play a central role in numerous cancers. In the recent years, both highly specific and broad-spectrum inhibitors of their catalytic activities have been successfully identified that are capable of increasing the sensitivity of tumor cells to cytotoxic agents. Because inhibiting DNA glycosylase activity in healthy cells could have dramatic consequences, alternative drug targets have also been identified that can modulate the activity of DNA glycosylases more specifically in tumor cells. Targeting the interactions between DNA glycosylases and their cellular partners that are enhanced in tumors, for example, constitutes a new and original approach, which has the advantage of not blocking DNA repair in normal, healthy cells, but instead to specifically inhibit the upregulation of DNA repair activity in drug-resistant tumor cells. DNA glycosylases are tightly regulated at the gene, mRNA, and protein levels, and in the future each one of these regulatory systems may represent a potent drug target to fine tune the activity of specific DNA glycosylases in a more personalized medicine approach.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21239226
References
- Stephen P. Jackson; Jiri Bartek; The DNA-damage response in human biology and disease. Nature 2009, 461, 1071-1078, 10.1038/nature08467.
- Jan H.J Hoeijmakers; Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366-374, 10.1038/35077232.
- Nimrat Chatterjee; Graham C. Walker; Mechanisms of DNA damage, repair, and mutagenesis. Environmental and Molecular Mutagenesis 2017, 58, 235-263, 10.1002/em.22087.
- Hans E. Krokan; Magnar Bjørås; Base Excision Repair. Cold Spring Harbor Perspectives in Biology 2013, 5, a012583-a012583, 10.1101/cshperspect.a012583.
- Nicholas C. Bauer; Anita H. Corbett; Paul W. Doetsch; The current state of eukaryotic DNA base damage and repair. Nucleic Acids Research 2015, 43, 10083-10101, 10.1093/nar/gkv1136.
- Miral Dizdaroglu; Erdem Coskun; Pawel Jaruga; Repair of oxidatively induced DNA damage by DNA glycosylases: Mechanisms of action, substrate specificities and excision kinetics. Mutation Research/Reviews in Mutation Research 2017, 771, 99-127, 10.1016/j.mrrev.2017.02.001.
- Miral Dizdaroglu; Oxidatively induced DNA damage and its repair in cancer. Mutation Research/Reviews in Mutation Research 2015, 763, 212-245, 10.1016/j.mrrev.2014.11.002.
- Susan S. Wallace; Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14-26, 10.1016/j.dnarep.2014.03.030.
- Muralidhar L. Hegde; Tapas K. Hazra; Sankar Mitra; Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Research 2008, 18, 27-47, 10.1038/cr.2008.8.
- Angelika L. Jacobs; Primo Schär; DNA glycosylases: in DNA repair and beyond. Chromosoma 2011, 121, 1-20, 10.1007/s00412-011-0347-4.
- Amanda K. McCullough; M. L. Dodson; R. Stephen Lloyd; Initiation of Base Excision Repair: Glycosylase Mechanisms and Structures. Annual Review of Biochemistry 1999, 68, 255-285, 10.1146/annurev.biochem.68.1.255.
- Miral Dizdaroglu; Base-excision repair of oxidative DNA damage by DNA glycosylases. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 2005, 591, 45-59, 10.1016/j.mrfmmm.2005.01.033.
- Harini Sampath; Amanda K. McCullough; R. Stephen Lloyd; Regulation of DNA glycosylases and their role in limiting disease. Free Radical Research 2012, 46, 460-478, 10.3109/10715762.2012.655730.
- Yun-Jeong Kim; David M. Wilson Iii; Overview of Base Excision Repair Biochemistry. Current Molecular Pharmacology 2012, 5, 3-13, 10.2174/1874-470211205010003.
- Thomas Hollis; Yoshitaka Ichikawa; Tom Ellenberger; DNA bending and a flip-out mechanism for base excision by the helix–hairpin–helix DNA glycosylase, Escherichia coli AlkA. The EMBO Journal 2000, 19, 758-766, 10.1093/emboj/19.4.758.
- M. M. Thayer; H. Ahern; D. Xing; R. P. Cunningham; John A Tainer; Novel DNA binding motifs in the DNA repair enzyme endonuclease III crystal structure.. The EMBO Journal 1995, 14, 4108-4120, 10.1002/j.1460-2075.1995.tb00083.x.
- Viswanath Bandaru; Sirisha Sunkara; Susan S. Wallace; Jeffrey P Bond; A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair 2002, 1, 517-529, 10.1016/s1568-7864(02)00036-8.
- Sylvie Doublié; Viswanath Bandaru; Jeffrey P. Bond; Susan S. Wallace; The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity. Proceedings of the National Academy of Sciences 2004, 101, 10284-10289, 10.1073/pnas.0402051101.
- Bjã¸rn Dalhus; Jon K. Laerdahl; Paul H. Backe; Magnar Bjørås; DNA base repair – recognition and initiation of catalysis. FEMS Microbiology Reviews 2009, 33, 1044-1078, 10.1111/j.1574-6976.2009.00188.x.
- Bret D. Freudenthal; Base excision repair of oxidative DNA damage from mechanism to disease. Frontiers in Bioscience 2017, 22, 1493-1522, 10.2741/4555.
- Hilde Nilsen; Marit Otterlei; Terje Haug; Kristin Solum; Toril A. Nagelhus; Frank Skorpen; Hans E. Krokan; Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Research 1997, 25, 750-755, 10.1093/nar/25.4.750.
- Antonio Sarno; Marie Lundbæk; Nina Beate Liabakk; Per Arne Aas; Robin Mjelle; Lars Hagen; Mirta M L Sousa; Hans E Krokan; Bodil Kavli; Uracil–DNA glycosylase UNG1 isoform variant supports class switch recombination and repairs nuclear genomic uracil. Nucleic Acids Research 2019, 47, 4569-4585, 10.1093/nar/gkz145.
- Joyce K. Hwang; Frederick W. Alt; Leng-Siew Yeap; Related Mechanisms of Antibody Somatic Hypermutation and Class Switch Recombination. Mobile DNA III 2015, 3, 325-348, 10.1128/microbiolspec.mdna3-0037-2014.
- Bas Pilzecker; Heinz Jacobs; Mutating for Good: DNA Damage Responses During Somatic Hypermutation. Frontiers in Immunology 2019, 10, 438, 10.3389/fimmu.2019.00438.
- Alfonso Bellacosa; Alexander C. Drohat; Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. DNA Repair 2015, 32, 33-42, 10.1016/j.dnarep.2015.04.011.
- Shinsuke Ito; Isao Kuraoka; Epigenetic modifications in DNA could mimic oxidative DNA damage: A double-edged sword. DNA Repair 2015, 32, 52-57, 10.1016/j.dnarep.2015.04.013.
- Richard B Roth; Leona D. Samson; 3-Methyladenine DNA glycosylase-deficient Aag null mice display unexpected bone marrow alkylation resistance.. Cancer Research 2002, 62, 656-660, .
- Aishwarya Prakash; Sylvie Doublié; Susan S. Wallace; The Fpg/Nei Family of DNA Glycosylases. Progress in Molecular Biology and Translational Science 2012, 110, 71-91, 10.1016/b978-0-12-387665-2.00004-3.
- Boris M. Brenerman; Jennifer L. Illuzzi; David M. Wilson; Base excision repair capacity in informing healthspan. Carcinogenesis 2014, 35, 2643-2652, 10.1093/carcin/bgu225.
- Jehad F. Alhmoud; John F. Woolley; Ala-Eddin Al Moustafa; Mohammed Imad Malki; DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050, 10.3390/cancers12041050.
- Luca Falzone; Salvatore Salomone; Massimo Libra; Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Frontiers in Pharmacology 2018, 9, 1300, 10.3389/fphar.2018.01300.
- A. Urruticoechea; R. Alemany; J. Balart; A. Villanueva; F. Vinals; G. Capella; Recent Advances in Cancer Therapy: An Overview. Current Pharmaceutical Design 2010, 16, 3-10, 10.2174/138161210789941847.
- Bruce A. Chabner; Thomas G. Roberts; Chemotherapy and the war on cancer. Nature Cancer 2005, 5, 65-72, 10.1038/nrc1529.
- Vincent T. DeVita; Edward Chu; A History of Cancer Chemotherapy. Cancer Research 2008, 68, 8643-8653, 10.1158/0008-5472.can-07-6611.
- Darío Galmarini; Carlos M. Galmarini; Felipe C. Galmarini; Cancer chemotherapy: A critical analysis of its 60 years of history. Critical Reviews in Oncology/Hematology 2012, 84, 181-199, 10.1016/j.critrevonc.2012.03.002.
- Grigory V. Mechetin; Anton V. Endutkin; Eugenia A. Diatlova; Dmitry O. Zharkov; Inhibitors of DNA Glycosylases as Prospective Drugs. International Journal of Molecular Sciences 2020, 21, 3118, 10.3390/ijms21093118.
- Torkild Visnes; Maurice Grube; Bishoy Magdy Fekry Hanna; Carlos Benitez-Buelga; Armando Cázares-Körner; Thomas Helleday; Targeting BER enzymes in cancer therapy. DNA Repair 2018, 71, 118-126, 10.1016/j.dnarep.2018.08.015.
- Dana Michel; Torkild Visnes; Evert J. Homan; Brinton Seashore-Ludlow; Mattias Hedenström; Elisée Wiita; Karl Vallin; Cynthia B. J. Paulin; Jiaxi Zhang; Olov Wallner; et al. Computational and Experimental Druggability Assessment of Human DNA Glycosylases. ACS Omega 2019, 4, 11642-11656, 10.1021/acsomega.9b00162.
- Inga R. Grin; Robert A. Rieger; Dmitry O. Zharkov; Inactivation of NEIL2 DNA glycosylase by pyridoxal phosphate reveals a loop important for substrate binding. Biochemical and Biophysical Research Communications 2010, 394, 100-105, 10.1016/j.bbrc.2010.02.121.
- Yngve T. Bliksrud; Amund Ellingsen; Magnar Bjørås; Fumarylacetoacetate inhibits the initial step of the base excision repair pathway: implication for the pathogenesis of tyrosinemia type I. Journal of Inherited Metabolic Disease 2012, 36, 773-778, 10.1007/s10545-012-9556-0.
- Artur Biela; Franck Coste; Françoise Culard; Martine Guerin; Stéphane Goffinont; Karola Gasteiger; Jarosław Cieśla; Alicja Winczura; Zygmunt Kazimierczuk; Didier Gasparutto; et al. Zinc finger oxidation of Fpg/Nei DNA glycosylases by 2-thioxanthine: biochemical and X-ray structural characterization. Nucleic Acids Research 2014, 42, 10748-10761, 10.1093/nar/gku613.
- Charlotte Rieux; Stéphane Goffinont; Franck Coste; Zahira Tber; Julien Cros; Vincent Roy; Martine Guérin; Virginie Gaudon; Stéphane Bourg; Artur Biela; et al. Thiopurine Derivative-Induced Fpg/Nei DNA Glycosylase Inhibition: Structural, Dynamic and Functional Insights. International Journal of Molecular Sciences 2020, 21, 2058, 10.3390/ijms21062058.
- Hao-Ching Wang; Chun-Han Ho; Chia-Cheng Chou; Tzu-Ping Ko; Ming-Fen Huang; Kai-Cheng Hsu; Andrew H.‐J. Wang; Using structural-based protein engineering to modulate the differential inhibition effects of SAUGI on human and HSV uracil DNA glycosylase. Nucleic Acids Research 2016, 44, 4440-4449, 10.1093/nar/gkw185.
- Suhman Chung; Jared B. Parker; Mario A Bianchet; L. Mario Amzel; James T. Stivers; Impact of linker strain and flexibility in the design of a fragment-based inhibitor. Nature Chemical Biology 2009, 5, 407-413, 10.1038/nchembio.163.
- Yu Lin Jiang; Daniel J. Krosky; Lauren Seiple; James T. Stivers; Uracil-Directed Ligand Tethering: An Efficient Strategy for Uracil DNA Glycosylase (UNG) Inhibitor Development. Journal of the American Chemical Society 2005, 127, 17412-17420, 10.1021/ja055846n.
- Daniel J. Krosky; Mario A. Bianchet; Lauren Seiple; Suhman Chung; L. Mario Amzel; James T. Stivers; Mimicking damaged DNA with a small molecule inhibitor of human UNG2. Nucleic Acids Research 2006, 34, 5872-5879, 10.1093/nar/gkl747.
- Aaron C. Jacobs; Marcus J. Calkins; Ajit Jadhav; Rjbal Dorjsuren; David Maloney; Anton Simeonov; Pawel Jaruga; Miral Dizdaroglu; Amanda K. McCullough; R. Stephen Lloyd; et al. Inhibition of DNA Glycosylases via Small Molecule Purine Analogs. PLOS ONE 2013, 8, e81667, 10.1371/journal.pone.0081667.
- Richard D. Kennedy; Clark C. Chen; Patricia Stuckert; Elyse M. Archila; Michelle A. De La Vega; Lisa A. Moreau; Akiko Shimamura; Alan D. D'andrea; Fanconi anemia pathway–deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. Journal of Clinical Investigation 2007, 117, 1440-1449, 10.1172/jci31245.
- Dorjbal Dorjsuren; Daemyung Kim; Vaddadi N. Vyjayanti; David J. Maloney; Ajit Jadhav; David M. Wilson; Anton Simeonov; Diverse Small Molecule Inhibitors of Human Apurinic/Apyrimidinic Endonuclease APE1 Identified from a Screen of a Large Public Collection. PLOS ONE 2012, 7, e47974, 10.1371/journal.pone.0047974.
- Aaron C. Jacobs; Marcus J. Calkins; Ajit Jadhav; Rjbal Dorjsuren; David Maloney; Anton Simeonov; Pawel Jaruga; Miral Dizdaroglu; Amanda K. McCullough; R. Stephen Lloyd; et al. Inhibition of DNA Glycosylases via Small Molecule Purine Analogs. PLOS ONE 2013, 8, e81667, 10.1371/journal.pone.0081667.
- Yu-Ki Tahara; Douglas Auld; Debin Ji; Andrew A. Beharry; Anna M. Kietrys; David L. Wilson; Marta Jimenez; Daniel King; Zachary Nguyen; Eric T. Kool; et al. Potent and Selective Inhibitors of 8-Oxoguanine DNA Glycosylase. Journal of the American Chemical Society 2018, 140, 2105-2114, 10.1021/jacs.7b09316.
- Muge Senarisoy; Caroline Barette; Françoise Lacroix; Salvatore De Bonis; Meike Stelter; Fabienne Hans; Jean-Philippe Kleman; Marie-Odile Fauvarque; Joanna Timmins; Förster Resonance Energy Transfer Based Biosensor for Targeting the hNTH1–YB1 Interface as a Potential Anticancer Drug Target. ACS Chemical Biology 2020, 15, 990-1003, 10.1021/acschembio.9b01023.
- Nathan Donley; Pawel Jaruga; Erdem Coskun; Miral Dizdaroglu; Amanda K. McCullough; R. Stephen Lloyd; Small Molecule Inhibitors of 8-Oxoguanine DNA Glycosylase-1 (OGG1). ACS Chemical Biology 2015, 10, 2334-2343, 10.1021/acschembio.5b00452.
- Bjørn Dalhus; Monika Forsbring; Ina Høydal Helle; Erik Sebastian Vik; Rune Johansen Forstrøm; Paul Hoff Backe; Ingrun Alseth; Magnar Bjørås; Separation-of-Function Mutants Unravel the Dual-Reaction Mode of Human 8-Oxoguanine DNA Glycosylase. Structure 2011, 19, 117-127, 10.1016/j.str.2010.09.023.
- Pietro Mancuso; Rossella Tricarico; Vikram Bhattacharjee; Laura Cosentino; Yuwaraj Kadariya; Jaroslav Jelinek; Emmanuelle Nicolas; Margret Einarson; Neil Beeharry; Karthik Devarajan; et al. Thymine DNA glycosylase as a novel target for melanoma. Oncogene 2019, 38, 3710-3728, 10.1038/s41388-018-0640-2.
- Torkild Visnes; Armando Cázares-Körner; Wenjing Hao; Olov Wallner; Geoffrey Masuyer; Olga Loseva; Oliver Mortusewicz; Elisée Wiita; Antonio Sarno; Aleksandr Manoilov; et al. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science 2018, 362, 834-839, 10.1126/science.aar8048.
- Uddhav K. Shigdel; Victor Ovchinnikov; Seung-Joo Lee; Jenny A. Shih; Martin Karplus; Kwangho Nam; Gregory L. Verdine; The trajectory of intrahelical lesion recognition and extrusion by the human 8-oxoguanine DNA glycosylase. Nature Communications 2020, 11, 4437, 10.1038/s41467-020-18290-2.
- Yu-Ki Tahara; Anna M. Kietrys; Marian Hebenbrock; Yujeong Lee; David L. Wilson; Eric T. Kool; Dual Inhibitors of 8-Oxoguanine Surveillance by OGG1 and NUDT1. ACS Chemical Biology 2019, 14, 2606-2615, 10.1021/acschembio.9b00490.
- Sarah K. Edwards; Toshikazu Ono; Shenliang Wang; Wei Jiang; Raphael M. Franzini; Jong Wha Jung; Ke Min Chan; Eric T. Kool; In Vitro Fluorogenic Real-Time Assay of the Repair of Oxidative DNA Damage. ChemBioChem 2015, 16, 1637-1646, 10.1002/cbic.201500184.
- Peter Sandin; Karl Börjesson; Hong Li; Jerker Mårtensson; Tom Brown; L. Marcus Wilhelmsson; Bo Albinsson; Characterization and use of an unprecedentedly bright and structurally non-perturbing fluorescent DNA base analogue. Nucleic Acids Research 2007, 36, 157-167, 10.1093/nar/gkm1006.
- Yusaku Nakabeppu; Eiko Ohta; Nona Abolhassani; MTH1 as a nucleotide pool sanitizing enzyme: Friend or foe?. Free Radical Biology and Medicine 2017, 107, 151-158, 10.1016/j.freeradbiomed.2016.11.002.
- Ashutosh Kumar; Tatsuro Kawamura; Makoto Kawatani; Hiroyuki Osada; Kam Y.J. Zhang; Identification and structure-activity relationship of purine derivatives as novel MTH1 inhibitors. Chemical Biology & Drug Design 2016, 89, 862-869, 10.1111/cbdd.12909.
- Debin Ji; Andrew A. Beharry; James M. Ford; Eric T. Kool; A Chimeric ATP-Linked Nucleotide Enables Luminescence Signaling of Damage Surveillance by MTH1, a Cancer Target. Journal of the American Chemical Society 2016, 138, 9005-9008, 10.1021/jacs.6b02895.
- Yusaku Nakabeppu; Eiko Ohta; Nona Abolhassani; MTH1 as a nucleotide pool sanitizing enzyme: Friend or foe?. Free Radical Biology and Medicine 2017, 107, 151-158, 10.1016/j.freeradbiomed.2016.11.002.
- Ashutosh Kumar; Tatsuro Kawamura; Makoto Kawatani; Hiroyuki Osada; Kam Y.J. Zhang; Identification and structure-activity relationship of purine derivatives as novel MTH1 inhibitors. Chemical Biology & Drug Design 2016, 89, 862-869, 10.1111/cbdd.12909.
- Debin Ji; Andrew A. Beharry; James M. Ford; Eric T. Kool; A Chimeric ATP-Linked Nucleotide Enables Luminescence Signaling of Damage Surveillance by MTH1, a Cancer Target. Journal of the American Chemical Society 2016, 138, 9005-9008, 10.1021/jacs.6b02895.