The PI3K/AKT/mTOR complex is a signaling pathway with a major role in essential cellular activities, such as: cell metabolism, cell growth, cell proliferation, apoptosis, and angiogenesis.
- breast cancer,PI3K/AKT/mTOR Signaling Pathway
1. PI3K/AKT/mTOR Signaling Pathway
Cells intercommunicate in a process called extracellular signaling. They produce specific molecules that bind to specific receptors of other cells and activate intracellular signaling pathways. This is how cells respond to changes and adapt [1].
The PI3K/AKT/mTOR complex is a signaling pathway with a major role in essential cellular activities, such as: cell metabolism, cell growth, cell proliferation, apoptosis, and angiogenesis [1]. A ligand (for instance insulin or an insulin-like growth factor) binds to a cell-membrane receptor (such as receptors for tyrosine kinases or G-protein-coupled-receptors-GPCR). The specific receptor, activated by the extracellular ligand, activates PI3K (phosphatidylinositol (3,4,5)-trisphosphate kinase). The activated PI3K catalyzes phosphorylation of PIP2 at the 3 position of the inositol ring to generate PIP3, which recruits two protein kinases to the plasma membrane via their pleckstrin homology interaction domains (PH domains): AKT (also called protein kinase B, or PKB) and PDK1 (phosphoinositide-dependent protein kinase 1). Once recruited to the cell membrane, the AKT is phosphorylated by mTORC2 (mTOR complex 2) on Ser473, changing the conformation of the AKT and allowing its phosphorylation on Thr308 by PDK1. The activated AKT phosphorylates target proteins from the cell membrane, then loses its connection with the cell membrane and phosphorylates other target proteins in the cytosol and cell nucleus. The phosphorylation of target proteins results in the stimulation of cell survival, growth, and proliferation [1].
2.1. PI3K/AKT/mTOR Signaling Pathway Members
2.1.1. PI3K-Phosphatidylinositol 3-Kinase-(Phosphoinositide 3-Kinase)
PI3K is a plasma-membrane-bound enzyme activated by RTKs (receptor tyrosine kinases) and by GPCRs (G protein-coupled receptors). GPCRs are the largest class of cellular surface receptors, with a generic structure; each GPCR is a transmembrane single polypeptide chain that uses G proteins to transmit the signal into the cytosol [1][2][3][4]. RTKs are a large family of plasma membrane receptors, too, with intrinsic protein kinase activity [2][3][4]. PI3Ks phosphorylate the 3’ position of the inositol head group of phosphatidylinositol (PIP2 and PIP3) lipids. PIP3 is the effector of multiple downstream targets of the phosphoinositide 3 kinase (PI3K) pathway [2][3][4]. There are many phosphatidylinositol 3-kinases-PI3Ks, divided into three groups or classes: PI3Ks class I, PI3Ks class II and PI3Ks class III [5].
PI3Ks class I is subdivided into PI3Ks class IA, PI3Ks class IB and PI3Ks class IC. IA-PI3Ks are heterodimers, consisting of a regulatory unit (p85α, p85β, p85γ) that activates the catalytic unit (p110α, p110β, p110δ, p110γ [5]). The IA-PI3Ks are activated directly by cell surface receptors: G protein-coupled receptors, RTKs and the small G protein RAS. Small GTPases form a superfamily within the larger class of regulatory GTP hydrolases, while RAS proteins are small GTPases that regulate cell growth, proliferation and differentiation [1][2][3][4]. IA-PI3Ks are present in many types of tissues and are activated by G protein- coupled receptors [5][6]. IB-PI3Ks are heterodimers containing the p101 regulatory subunit, which activates the p110γ catalytic subunit [7][8].
Class II PI3Ks has three isoforms: PI3KC2α and PI3KC2β are expressed in most of the tissues and organs, while PI3KC2γ is expressed only in the liver [9]. They regulate intracellular membrane dynamics and membrane traffic [9][10]. Class III PI3Ks has only one member identified: VPS34, which is connected to regulation of phagocytosis, pinocytosis, endosomal sorting and autophagy [11].
2.1.2. AKT
The serine/threonine protein kinase AKT is the principal downstream molecule of the PI3K signaling pathway. There are three subtypes (isoforms) of AKT [12][13]: AKT1 (expressed in the majority of tissues), AKT2 (expressed mainly in tissues with high sensitivity to insulin: liver, pancreas, muscles), and AKT3 (expressed in the brain and testicles). AKT is activated by PIP2-driven and PIP3-driven recruitment to the plasma membrane. Here, the phosphorylation of Thr308 and of Ser473 determines the activation of AKT [14][15].
Activated AKT mediates the regulation of the cell cycle, growth, proliferation, and energy metabolism [16]. AKT has over 100 substrates [12], including: transcription factors, inhibitors of cell cycle progression, protein kinases, GTPase-activation proteins, and apoptosis inducers [17][18].
Glycogen synthase kinase-3 (GSK-3), one of the main AKT protein substrates, is a protein kinase that phosphorylates and inhibits the glycogen synthase. GSK-3 lies downstream of multiple cellular signaling pathways, such as: the phosphatidylinositol-3–kinase-dependent pathway that is stimulated by insulin and growth factors, and the Wnt signaling pathway that is required for embryonic development [19]. GSK-3 is primarily regulated by inhibition [20]. There are two isoforms, GSK-3α and GSK-3β, generated from distinct genes, but with great structural homology (almost 97%) and similar roles, being encountered in many tissues [19][20] and especially in the brain. AKT phosphorylates GSK-3 and inactivates it; consequently, there is an increase in the cellular uptake of glucose and glycogen synthesis. This determines a decrease of blood sugar levels [21].
The inhibition of GSK-3 triggered by growth factors, through AKT activation, has anti-apoptotic effects [20]. GSK-3 has a broad range of substrates (more than 100) including signaling proteins, structural proteins, and transcription factors involved in metabolism [22].
2.1.3. mTOR
TOR is a large protein-kinase inactivated by a bacterial toxin called Rapamycin—hence the name Target of Rapamycin. It was identified in yeasts, but it also exists in mammalian cells, being named mTOR (mammalian Target of Rapamycin) [1][23]. In cells, it exists as two distinct multiprotein complexes: mTORC1 and mTORC2. mTORC1 contains mTOR, protein Raptor and mLST8 (the acronym for mammalian Lethal with SEC13 protein 8). mLST8 interacts directly with mTOR and enhances its kinase activity, with this protein being found in human colon and prostate cancer cells [24]. mTORC1 is sensitive to Rapamycin and promotes cell growth and survival by stimulating nutrient uptake and metabolism [1][23]. It also stimulates cell growth by promoting ribosome production and protein synthesis and by inhibiting protein degradation [1][23]. mTORC1 may be activated through different pathways, but mainly through the PI3P/AKT pathway, which is activated by extracellular growth factors and nutrients. Activated AKT phosphorylates the Tuberous Sclerosis protein 2 (TSC2), which becomes inactive. Thus, TSC2 cannot keep Rheb (a Ras-related GTPase) in its inactive form. Consequently, Rheb-GDP (inactive) becomes Rheb-GTP (active), contributing to the activation of mTORC1 [1][23][25]. Other targets of the mTORC1 are: S6K (a protein kinase that phosphorylates the ribosomal protein S6) and 4E-BP (an inhibitor of the translation initiation factor eIF4E); the consequences are increased production of ribosomes and increased protein synthesis [25].
mTORC2 consists of mTOR, protein Rictor, Sin1 and mLST1 and is not sensitive to Rapamycin. mTORC2 promotes AKT activation by directly phosphorylating its hydrophobic motif (Ser473). This permits further phosphorylation of AKT, at Thr308, by PDK1 and so AKT becomes fully active [1][23]. Sin1 contains a phospholipid-binding pleckstrin homology (PH) domain that facilitates the association of mTORC2 with membranes [26]. Phosphorylation of Sin1 at Thr86 and Thr398 (by S6K or AKT) dissociates Sin1 from mTORC2, thus resulting mTORC2 inhibition[27]. Acting on S6K, mTORC1 directly regulates mTORC2[27]. mTORC2 is mainly involved in the reconstruction of the cytoskeleton (through the Rho family GTPases) and cell survival [1].
2.1.4. FoxO1
Forkhead box other 1 is a member of the Forkhead transcription factor family. The family is divided into 17 subfamilies named FoxA to FoxQ [28]. There is a common feature of the Forkhead family, namely a conserved DNA-binding domain called Fox [29].
FoxO1 is important for the glucose and lipids’ metabolism. It enhances the synthesis of enzymes involved in gluconeogenesis, has a suppressive effect on the synthesis of enzymes of glycolysis, inhibits the pentose phosphate pathway, and diminishes the triacylglycerol synthesis. Insulin activates the RTKs and initiates the PI3P/AKT pathway. The activated AKT phosphorylates the FoxO1 existing in the cytosol. The phosphorylated FoxO1 is tagged by the attachment of ubiquitin and is then degraded by proteasomes. The unphosphorylated FoxO1 remains active, passes from the cytosol into the nucleus, binds to a response element, and triggers the transcription of its associated genes, such as PEP-carboxykinase, glucose 6-phosphatase, etc. FoxO family members have an important role in oxidative stress resistance, cell proliferation, apoptosis, and differentiation [30][31].
2.1.5. PTEN
PTEN (phosphatase and tensin homolog) is a PIP3 specific phosphatase that dephosphorylates the PIP3 molecules, resulting in PIP2 molecules (Figure 1). PIP2 is not a binding dock for AKT, so AKT cannot be recruited to the cell membrane. As a consequence, AKT cannot be phosphorylated by mTORC2 on Ser473; therefore, the conformation of the AKT does not change anymore and the phosphorylation on Thr308 by PDK1 is not permitted. The result is that AKT cannot be activated and the PI3K/AKT/mTOR signaling pathway is suppressed [32][33]. That is why PTEN acts as a tumor suppressor, by inhibiting cell proliferation [32][33]. In many malignant tumors, the PTEN gene has suffered mutations, resulting in abnormal PTEN, which cannot exert its inhibitory effect on the PIP3/AKT/mTOR pathway [34]. The plasmatic levels of PIP3 rise and the activity of AKT is continuously stimulated [35]. By modulating the PIP3/AKT/mTOR pathway, PTEN is linked to glucose homeostasis [36].
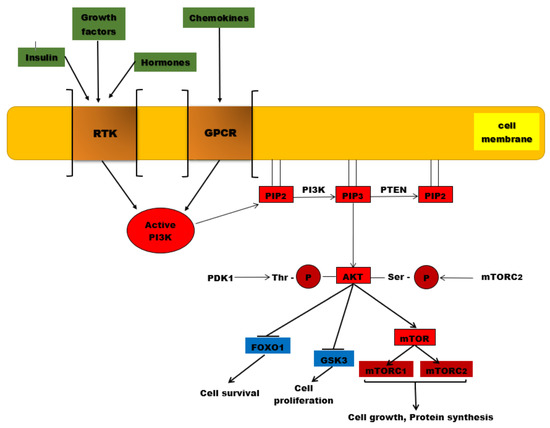
Figure 1. The PI3K/AKT/mTOR signaling pathway. PI3K is activated by the binding of ligands (insulin, growth factors, hormones) to RTKs, but also to GPCR (chemokines). Once activated, this protein kinase will catalyze the phosphorylation of PIP2 to PIP3. AKT is recruited to the plasma membrane where it undergoes two phosphorylation processes, one catalyzed by PDK1 at the level of threonine residue and the second reaction being catalyzed by mTORC2. Once activated by phosphorylation, AKT will phosphorylate other substances such as the mTOR complex, which will be associated in the end with protein synthesis and cell growth. Other phosphorylated substrates, such as GSK-3 and Fox01, will be inhibited, associated with cell proliferation and survival. PTEN is the major negative regulator of this signaling pathway involved in PIP3 dephosphorylation.
3. PI3K/AKT/mTOR Mutations in Breast Cancer
The PI3K pathway undergoes many changes in breast cancer caused by mutations or amplifications of genes which encode the catalytic subunits p110α (PIK3CA) and p110 β (PIK3CB), but also the regulatory subunit PI3K, p85α (PIK3R1) [37]. In human neoplasms, PIK3CA is the frequently mutated gene that encodes the p110α catalytic subunit of the PI3K pathway, and was found amplified in head and neck, cervical, gastric, lung and breast cancers. In prostate, breast, endometrium and colon cancers, the highest incidence of PIK3CA mutations has been detected [38].
Approximately 30–40% of patients with breast cancer present PIK3CA mutations, which will induce hyperactivation of the α isoform (p110α) of PI3K. Recently, the FDA (Food and Drug Administration) approved testing of breast cancer patients with PIK3CA mutations using breast tumor tissue and/or circulating tumor DNA, isolated from plasma specimens. The results reported 11 PIK3CA hotspots mutations, located mainly in exons 9 and 20. Gene PIK3CA mutations have been detected using a PCR test, with the results revealing the following exon mutations—exon 9: E542K, E545A, E545D, E545G, E545K, Q546E, and Q546R; and exon 20: H1047L, H1047R, and H1047Y [39]. PI3Kα is activated both by binding insulin or growth factors to RTKs and by oncogenic mutations [40]. In breast cancer, the PI3K/AKT pathway is activated through PIK3CA or AKT1 mutations and PTEN loss [41].
In 2004, Samuels Y and co-workers reported somatic mutations of PIK3CA coding p110α in various solid malignancies for the first time[42]. Samuels Y et al., observed that the majority of PIK3CA somatic mutations are located at the level of exon 9 (E542K or E545K) and exon 20 (H1047R or H1047L). In the helical domain of p110α, there are exon 9 mutations that are considered to enable p110α to escape the inhibitory effect of p85 via the Src-homology 2 (SH2) domain. Near the activation loop of the kinase domain, mutations of exon 20 are located. The study reported 10% frequency of PIK3CA somatic mutations in breast cancer, but later studies reported ∼30%. [43]. Karakas B et al., also reported that the catalytic subunit of the PI3K gene called PIK3CA or p110α is frequently mutated in breast cancer [44].
In the coding sequence, PIK3CA mutations are concentrated in three hotspots, with two located in the helical domain of p110α and the last situated in the catalytic domain. The hotspot mutations represent the single nucleotide substitutions that will determine amino acid substitutions, E542 K, E545 K and H1047R. Unfortunately, these hotspot mutations induce a gain-of-function and prompt transformation and tumorigenicity. The results from 6338 tumors revealed that 2261 patients presented PIK3CA mutations (35.7%) [45].
A total of 73% of all PIK3CA mutations are: H1047R (35%), E545K (17%), E542K (11%), N345K (6%), and H1047L (4%). In patients with triple negative breast cancer, PIK3CA mutation rates were decreased (16%) compared to HR+/HER2 (42%) and HER2+ (31%) breast cancer subtypes. Moreover, in patients with advanced HR+/HER2−breast cancer, 28% of PIK3CA mutations were identified in circulating tumor DNA [39].
Tumor sequencing studies have reported that these somatic mutations of PI3CA, concentrated in certain hotspots, will lead to tumor progression by gaining a function for PI3CA [46]. Moreover, PIK3CA mutations in human breast cancers, at E545K in exon 9 and H1047R in exon 20, have been reported even by studies using cell lines such as MCF10A immortalized breast epithelial cells. PIK3CA was the most frequent mutation observed, associated with an increased kinase activity of the PI3K pathway. Mutant PIK3CA promotes cell growth and invasion of human cancer cells [43].
Bachman KE and co-workers reported an incidence of 25% PIKCA mutation in human breast cancer. The study did not reveal any correlation between PIK3CA and the presence or absence of ER/PR labelling, or even with Her-2/neu. PI3CA mutations affect the PI3K/AKT/mTOR signaling pathway independent of ER/PR and Her-2/neu. Analyzing the fifty-three samples, the study reported three mutations in exon 9, 8 uncovered mutations in exon 20, and novel somatic mutations were detected—two in exon 1 and one in exon 2 [47]. Stemke-Hale K and colleagues analyzed 547 breast tumor samples and 41 cell lines using mass spectrometry sequencing and reverse-phase protein arrays to detect mutations in PI3KCA, AKT and PTEN. The study revealed that the most common PIK3CA mutations were found in hormone receptor-positive forms (34.5%) followed by HER2- positive cases with an incidence of 22.7%, compared with basal-like tumors (8.3%). Moreover, in hormone receptor-positive cancers, mutations on AKT1 represented 1.4% and PTEN 2.3%, respectively [48]. Using cell cultures, the study reported that AKT1 mutations were absent, while PIK3CA and PTEN mutations appeared in 39% and 20% of the cases, respectively. In tumors and cell lines, PIK3CA mutations compared with the loss of PTEN and AKT1 mutations were associated with less activation of AKT. The most frequent modifications on the PI3K/AKT/mTOR signaling pathway were PTEN loss and PIK3CA mutation [48].
Li SY. et al., analyzed 250 primary human breast tumors and detected that 35% of PIK3CA mutations were located in C2 helical and kinase domains. The PIK3CA mutations were associated with larger tumors and significantly worse survival rate, especially in positive estrogen receptor status or non-amplified ERBB2 [49]. Moreover, PIK3CA mutations may sometimes harbor PTEN loss or HER2 overexpression in breast tumors [47].
p110α, the catalytic subunit of the phosphoinositide 3-kinase alpha (PI3Kα) complex, which is necessary for normal growth and proliferation, [45] is essential for signaling and the growth of tumors driven by PIK3CA mutations or RTKs [43]. It has been shown that p110β mediates tumorigenesis in PTEN-deficient cells [49]. Breast cancers show poor disease outcome if they are associated with increased levels of AKT phosphorylation/activation and PTEN loss. Moreover, the loss of PTEN activity and activation of the PI3K signaling pathway are associated with resistance to endocrine therapy [36]. Endometrial, prostate, breast, thyroid and kidney tumors present somatic PTEN alterations, leading to uncontrolled PI3K activation [50]. PTEN, the most important regulator of the PI3K/AKT/mTOR signaling pathway, is involved in cell growth and survival, cellular migration and genomic stability. In 1997, it was discovered that PTEN acts as a key tumor suppressor gene for various tumor types, being involved in cell cycle progression, cell growth and survival. Moreover, PTEN is implicated in DNA repair and genome stability. In response to DNA damage, PTEN is phosphorylated (Tyr) and binds to chromatin, promoting DNA repair [51].
The somatic mutations (missense and nonsense mutations, monoallelic or biallelic deletion on the PTEN gene), epigenetic alterations (methylation promotor), PTEN protein degradation and the post-translational modification of PTEN protein will conduce to PTEN inactivation. In breast tumors, the loss of heterozygosity at the PTEN locus was detected in 40–50% cases. The loss of PTEN function due to PTEN mutations is found in 5–10% of breast cancers [51].
In luminal breast cancers, the PI3K pathway is one of the most altered pathways, correlated with PIK3CA mutations, loss of PTEN, or downstream protein phosphorylation . Zardavas D et al., reported the results obtained from 10,319 patients included in 19 studies where PIK3CA mutations were present in 32% of patients. PIK3CA mutations were associated with ER positivity, and were increased with age, lower grade, and smaller size. In breast cancer subtypes-ER-negative/HER2-negative, HER2-positive, and ER-positive/HER2-negative, the prevalence of PIK3CA mutations was 18%, 22%, and 37%, respectively [52]. Ling D et al., conducted a study in which tumors from 507 breast cancer patients were collected from the West China Hospital between 2008 and 2013. The study’s results revealed 3.% AKT1 mutations with ER+/PR+/HER2. The incidence of the PIK3CA mutations was reported at 46.5%. These mutations were associated with ER+/PR+/HER2‒ status, and it was observed that 35 patients carried two or three variants of the PIK3CA gene [53]. PIK3CA mutations, associated with many distinct cancers, include hotspot single–amino acid substitutions in the helical (E542K and E545K) or kinase (H1047R) domains. In multiple cancer types, including breast cancer, PIK3CA is considered oncogenic, mutations of the alpha catalytic subunit of PI3K having an incidence of 40% in ER+/HER2− primary and metastatic tumors. Therefore, PIK3CA is a target for cancer therapy [48]. Anderson EJ et al., reported 36% PIK3CA mutation in HR+/HER2- metastatic breast cancer [54].
Mutations also occur in RTKs such as HER2 (ERBB2) and fibroblast growth factor receptor (FGFR)1, in AKT1, AKT2, PDK1, and loss of PTEN and INPP4B (inositol polyphosphate-4-phosphatase type II). The activation of PI3K occurs through the binding of growth factors to RTKs and GPCR. Moreover, PIK3CA mutations appear in breast tumors associated with PTEN loss or HER overexpression [28].
Lehmann BD and co-workers detected highly clonal PIK3CA mutations in the triple negative breast cancer subtype that present a luminal phenotype and express androgen receptors (40%) versus triple negative breast cancer without androgen receptors (4%) [55]. A total of 15–20% of breast cancer cases present an overexpression of human epidermal growth factor receptor-2 (HER2), associated with an aggressive clinical behavior [56]. Luminal A tumors are associated with PIK3CA mutations in 45% of the cases, while AKT1 and PTEN mutations both appear in 4% of the patients. At the same time, PIK3CA genes are mutated in 29% of the cases with subtype luminal B, in 39% of HER2-enriched breast cancers and only in 7% of basal-like tumors [57].
HER Receptors and Breast Cancers
EGFR (epidermal growth factor receptor, also known as ERBB1/HER1), ERBB2 (HER2), ERBB3 (HER3), and ERBB4 (HER4) represent the ERBB family of RTKs, which are cytoplasmic membrane-anchored proteins. All four receptors display similarities in structure and sequence, contain an extracellular ligand-binding domain, a transmembrane domain, and an intracellular tyrosine kinase domain [58][152].
Cell growth, survival, and differentiation are regulated by HER receptors via various signaling pathways and even participate in cellular proliferation and differentiation. The HER2 gene encodes a 185-kDa transmembrane protein, being located on the long arm of chromosome 17 [59]. When HER2 is overexpressed or amplified, it stimulates tumor growth, invasiveness, and survival via the activation of several signaling cascades, such as PI3K/AKT pathways. HER2 phosphorylation may lead to PI3K/AKT/mTOR pathway activation [60].
The formation of HER2-EGFR dimers, HER2 homodimers and even HER2-HER3 dimers will promote tumor development by increasing tumor cell metabolic functions, cell survival, proliferation and invasiveness [61]. In breast cancer, overactivation of HER receptors is caused by several factors such as gene amplification, truncation of the extracellular domain, mutations in the kinase domain, and co-expression of HER receptor ligands [62]. HER2 overexpression is associated with poor clinical outcome and disease progression [58].
In primary invasive breast cancer, approximately 18–20% of cases present an amplification or overexpression of the HER2 oncogene [60]. HER 2 (c-erbB-2) is a cell membrane surface-bound RTK, while HER2/neu, its extracellular domain, is normally implicated in the signal transduction pathways that will conduce to cell growth and differentiation. In approximately 15–20% of breast cancer cases, HER2-overexpression was observed [63]. HER2 overexpression and PIK3CA mutations have been observed in both invasive breast cancers and ductal carcinoma in situ. In intraepithelial neoplastic lesions, PIK3CA mutations have a decreased frequency, so these mutations can enhance PI3K pathway activation by HER 2 (ERBB2) [43].
EGFR, HER3, and HER4 are amplified and overexpressed in more than 20% of breast cancers. Moreover, HER2 is the oncogenic driver of these pathologies, involved in the genesis and progression of these tumors [64]. EGFR and HER4 can activate PI3K after their binding to RTKs, especially by transphosphorylation of HER3, which can act as a critical partner for HER2 in the genesis and progression of the tumor [64]. After phosphorylation of tyrosine residues within the cytoplasmic domain, dimerization of the receptor takes place and various signaling pathways are activated, which are further involved in cellular proliferation, transcription, motility, and inhibition of apoptosis [60].
Yang Z et al., conducted a study that included 142 patients with metastatic breast cancer, detecting alterations in estrogen receptor (ER), progesterone receptor (PR), and HER2 status as follows: 20.70%, 37.78%, and 11.48%, respectively [65].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22010173
References
- Lim, W.; Mayer, B.; Pawson, T. Cell Signaling: Principles and Mechanisms; Garland Science: New York, NY, USA, 2015.
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–384. [Google Scholar] [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: A conserved switch for diverse cell functions. Nature 1990, 348, 125–132. [Google Scholar] [CrossRef]
- Paduch, M.; Jelen, F.; Otlewski, J. Structure of small G proteins and their regulators. Acta Biochim. Pol. 2001, 48, 829–850. [Google Scholar] [CrossRef]
- Yudushkin, I. Getting the Akt together: Guiding intracellular Akt activity by PI3K. Biomolecules 2019, 9, 67. [Google Scholar] [CrossRef]
- Yu, X.; Long, Y.C.; Shen, H.M. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy 2015, 11, 1711–1728.
- Lehninger, A.; Nelson, D.L.; Cox, M.C.; Freeman, W.H. Lehninger Principles of Biochemistry; W.H. Freeman: New York, NY, USA, 2012. [Google Scholar]
- Balla, T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef] [PubMed]
- Braccini, L.; Ciraolo, E.; Campa, C.C.; Perino, A.; Longo, D.L.; Tibolla, G.; Pregnolato, M.; Cao, Y.; Tassone, B.; Damilano, F.; et al. PI3K-C2γ is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat. Commun. 2015, 6, 7400. [Google Scholar] [CrossRef] [PubMed]
- Falasca, M.; Hughes, W.E.; Dominguez, V.; Sala, G.; Fostira, F.; Fang, M.Q.; Cazzolli, R.; Shepherd, P.R.; James, D.E.; Maffucci, T. The role of phosphoinositide 3-kinase C2α in insulin signaling. J. Biol. Chem. 2007, 282, 28226–28236. [Google Scholar] [CrossRef] [PubMed]
- Backer, J. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem. J. 2016, 473, 2251–2271. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB signaling: Navigating the network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Dummler, B.; Hemmings, B.A. Physiological roles of PKB/Akt isoforms in development and disease. Biochem. Soc. Trans. 2007, 35, 231–235. [Google Scholar] [CrossRef]
- Szymonowicz, K.; Oeck, S.; Malewicz, N.M.; Jendrossek, V. New insights into protein kinase B/Akt signaling: Role of localized Akt activation and compartment-specific target proteins for the cellular radiation response. Cancers 2018, 10, 78. [Google Scholar] [CrossRef]
- Revathidevi, S.; Munirajan, A.K. Akt in cancer: Mediator and more. Semin. Cancer Biol. 2019, 59, 80–91. [Google Scholar] [CrossRef]
- Risso, G.; Blaustein, M.; Pozzi, B.; Mammi, P.; Srebrow, A. Akt/PKB: One kinase, many modifications. Biochem. J. 2015, 468, 203–214. [Google Scholar] [CrossRef]
- Luo, C.T.; Li, M. Foxo transcription factors in T cell biology and tumor immunity. Semin. Cancer Biol. 2018, 50, 13–20. [Google Scholar] [CrossRef]
- Arcaro, A.; Guerreiro, A.S. The phosphoinositide 3-kinase pathway in human cancer: Genetic alterations and therapeutic implications. Curr. Genom. 2007, 8, 271–306. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Woodgett, J.R. Glycogen Synthase Kinase 3: A Kinase for All Pathways? Curr. Top. Dev. Biol. 2017, 123, 277–302. [Google Scholar] [PubMed]
- Dokken, B.B.; Sloniger, J.A.; Henriksen, E.J. Acute selective glycogen synthase kinase-3 inhibition enhances insulin signaling in prediabetic insulin-resistant rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1188–E1194. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, P.A.; Coghlan, M.; Rice, S.Q.; Sutherland, C. Inhibition of GSK-3 selectively reduces glucose-6-phosphatase and phosphatase and phosphoenolypyruvate carboxykinase gene expression. Diabetes 2001, 50, 937–946. [Google Scholar] [CrossRef]
- Wei, X.; Luo, L.; Chen, J. Roles of mTOR signaling in tissue regeneration. Cells 2019, 8, 1075. [Google Scholar] [CrossRef]
- Kakumoto, K.; Ikeda, J.; Okada, M.; Morii, E.; Oneyama, C. mLST8 promotes mTOR-mediated tumor progression. PLoS ONE 2015, 10, e0119015. [Google Scholar] [CrossRef]
- Mahoney, R.E.; Azpurua, J.; Eaton, B.A. Insulin signaling controls neurotransmission via the 4eBP-dependent modification of the exocytotic machinery. eLife 2016, 5, e16807. [Google Scholar] [CrossRef]
- Berchtold, D.; Walther, T.C. TORC2 Plasma membrane localization is essential for cell viability and restricted to a distinct domain. Mol. Biol. Cell 2009, 20, 1565–1575. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Inuzuka, H.; Lazorchak, A.S.; Gao, D.; Arojo, O.; Liu, D.; Wan, L.; Zhai, B.; Yu, Y.; et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat. Cell Biol. 2013, 15, 1340–1350. [Google Scholar] [CrossRef]
- Hollenhorst, P.C.; Bose, M.E.; Mielke, M.R.; Müller, U.; Fox, C.A. Forkhead genes in transcriptional silencing, cell morphology and the cell cycle. Overlapping and distinct functions for FKH1 and FKH2 in Saccharomyces cerevisiae. Genetics 2000, 154, 1533–1548. [Google Scholar]
- Cabrera-Ortega, A.; Feinberg, D.; Liang, Y.; Rossa, J.C.; Graves, D.T. The role of Forkhead Box 1 (FOXO1) in the immune system: Dendritic cells, T cells, B cells, and hematopoietic stem cells. Crit. Rev. Immunol. 2017, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Xin, Z.; Hu, W.; Jiang, S.; Yang, Z.; Yan, X.; Li, X.; Yang, Y.; Chen, F. Forkhead box O proteins: Crucial regulators of cancer EMT. Semin. Cancer Biol. 2018, 50, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K. Forkhead transcription factors: Formulating a FOXO target for cognitive loss. Curr. Neurovascular Res. 2017, 14, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Cretella, D.; Digiacomo, G.; Giovannetti, E.; Cavazzoni, A. PTEN alterations as a potential mechanism for tumor cell escape from PD-1/PD-L1 inhibition. Cancers 2019, 11, 1318. [Google Scholar] [CrossRef]
- Luongo, F.; Colonna, F.; Calapà, F.; Vitale, S.; Fiori, M.E.; De Maria, R. PTEN tumor-suppressor: The dam of stemness in cancer. Cancers 2019, 11, 1076. [Google Scholar] [CrossRef]
- Naderali, E.; Khaki, A.A.; Rad, J.S.; Alihemmati, A.; Rahmati, M.; Nozad-Charoudeh, H. Regulation and modulation of PTEN activity. Mol. Biol. Rep. 2018, 45, 2869–2881. [Google Scholar] [CrossRef]
- Maehama, T.; Taylor, G.S.; Dixon, J.E. PTEN and myotubularin: Novel phosphoinositide phosphatases. Annu. Rev. Biochem. 2001, 70, 247–279. [Google Scholar] [CrossRef]
- Nguyen, K.T.; Tajmir, P.; Lin, C.H.; Liadis, N.; Zhu, X.D.; Eweida, M.; Tolasa-Karaman, G.; Cai, F.; Wang, R.; Kitamura, T.; et al. Essential role of PTEN in body size determination and pancreatic beta-cell homeostasis in vivo. Mol. Cell. Biol. 2006, 26, 4511–4518. [Google Scholar] [CrossRef]
- Abraham, J. PI3K/AKT/mTOR pathway inhibitors: The ideal combination partners for breast cancer therapies? Expert Rev. Anticancer Ther. 2015, 15, 51–68. [Google Scholar] [CrossRef]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224. [Google Scholar] [CrossRef]
- Chalhoub, C.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sáez, O.; Chic, N.; Pascual, T.; Adamo, B.; Vidal, M.; González-Farré, B.; Sanfeliu, E.; Schettini, F.; Conte, B.; Brasó-Maristany, F.; et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 2020, 22, 45. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Razavi, P.; Johnson, J.L.; Shao, H.; Shah, H.; Antoine, A.; Ladewig, E.; Gorelick, A.N.; Lin, T.-Y.; Toska, E.; et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors. Science 2019, 366, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.A.; Yisheng, L.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 78. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Ptak, N.J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; Willson, J.K.V.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Diaz, L.A.; Schmidt-Kittler, O.; Cummins, J.M.; Delong, L.; Cheong, I.; Rago, C.; Huso, D.L.; Lengauer, C.; Kinzler, K.W.; et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005, 7, 561–573. [Google Scholar] [CrossRef]
- Karakas, B.; Bachman, K.E.; Park, B.H. Mutation of the PIK3CA oncogene in human cancers. Br. J. Cancer 2006, 94, 455–459. [Google Scholar] [CrossRef]
- Rand, A.; Yardena, S. PIK3CA in cancer: The past 30 years. Semin. Cancer. Biol. 2019, 59, 36–49. [Google Scholar]
- Saal, L.H.; Holm, K.; Maurer, M.; Memeo, L.; Su, T.; Wang, X.; Yu, J.S.; Malmström, P.O.; Mansukhani, M.; Enoksson, J.; et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005, 1, 2554–2559. [Google Scholar] [CrossRef]
- Bachman, K.E.; Argani, P.; Samuels, Y.; Silliman, N.; Ptak, J.; Szabo, S.; Konishi, H.; Karakas, B.; Blair, B.G.; Lin, C.; et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol. Ther. 2004, 3, 772–775. [Google Scholar] [CrossRef]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.-L.; Davies, M.; Carey, M.; Yinghui, G.; Guan, Y.; Sahin, A.; et al. An Integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008, 68, 6084–6091. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Rong, M.; Grieu, F.; Iacopetta, B. PIK3CA mutations in breast cancer are associated with poor outcome. Breast Cancer Res. Treat. 2005, 96, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.-H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Carbognin, L.; Miglietta, F.; Paris, I.; Dieci, M.V. Prognostic and predictive implications of PTEN in breast cancer: Unfulfilled promises but intriguing perspectives. Cancers 2019, 11, 1401. [Google Scholar] [CrossRef] [PubMed]
- Zardavas, D.; Te Marvelde, L.; Milne, R.L.; Fumagalli, D.F.; Fountzilas, G.; Kotoula, V.; Razis, E.; Papaxoinis, G.; Joensuu, H.; Moynahan, M.E.; et al. Tumor PIK3CA genotype and prognosis in early-stage breast cancer: A pooled analysis of individual patient data. J. Clin. Oncol. 2018, 1, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Ling, D.; Xuehua, Z.; Yun, S.; Jiemin, W.; Xiaorong, Z.; Jiayuan, L.; Min, H.; Hong, Z. Prevalence and prognostic role of PIK3CA/AKT1 mutations in chinese breast cancer patients. Cancer. Res. Treat. 2019, 51, 128–140. [Google Scholar]
- Anderson, A.J.; Mollon, L.E.; Dean, J.L.; Warholak, T.L.; Aizer, A.; Platt, E.A.; Tang, D.H.; Lisa, E.; Davis, L.E. A systematic review of the prevalence and diagnostic workup of PIK3CA mutations in HR+/HER2− metastatic breast cancer. Int. J. Breast. Cancer 2020, 2020, 3759179. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Schafer, J.M.; Pendleton, C.S.; Tang, L.; Johnson, K.C.; Chen, X.; Balko, J.M.; Gómez, H.L.; Arteaga, C.L.; et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014, 8, 406. [Google Scholar] [CrossRef]
- Dieci, M.V.; Miglietta, F.; Griguolo, G.; Guarneri, V. Biomarkers for HER2-positive metastatic breast cancer: Beyond hormone receptors. Cancer Treat. Rev. 2020, 88, 102064. [Google Scholar] [CrossRef]
- Toss, A.; Cristofanilli, M. Molecular characterization and targeted therapeutic approaches in breast cancer. Breast Cancer Res. 2015, 17, 60. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.H.; Hung, M.C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Mitri, Z.; Constantine, T.; O’Regan, R. The HER2 receptor in breast cancer: Pathophysiology, clinical use, and new advances in therapy. Chemother. Res. Pract. 2012, 2012, 743193. [Google Scholar] [CrossRef] [PubMed]
- Gagliato, D.D.M.; Jardim, D.L.F.; Marchesi, M.S.P.; Hortobagyi, G.N. Mechanisms of resistance and sensitivity to anti-HER2 therapies in HER2+ breast cancer. Oncotarget 2016, 7, 64431–64446. [Google Scholar] [CrossRef] [PubMed]
- Schettini, F.; Buono, G.; Cardalesi, C.; Desideri, I.; De Placido, S.; Del Mastro, L. Hormone receptor/human epidermal growth factor receptor 2-positive breast cancer: Where we are now and where we are going. Cancer Treat. Rev. 2016, 46, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Babak, N.; Hamid, M.; Zhixiang, W. Mechanisms underlying the action and synergism of trastuzumab and pertuzumab in targeting HER2-positive breast cancer. Cancers 2018, 10, 342. [Google Scholar]
- Kechagioglou, P.; Papi, R.; Provatopoulou, X.; Kalogera, E.; Papadimitriou, E.; Grigoropoulos, P.; Nonni, A.; Zografos, G.; Kyriakidis, D.A.; Gounaris, A. Tumor suppressor PTEN in breast cancer: Heterozygosity, mutations and protein expression. Anticancer Res. 2014, 34, 1387–1400. [Google Scholar]
- Ruiz-Saenz, A.; Dreyer, C.; Campbell, M.R.; Steri, V.; Gulizia, N.; Moasser, M. HER2 amplification in tumors activates PI3K/AKT signaling independent of HER3. Cancer Res. 2018, 78, 3655–3658. [Google Scholar] [CrossRef]
- Yang, Z.; Li, N.; Li, X.; Lei, L.; Wang, X. The prognostic impact of hormonal receptor and HER-2 expression discordance in metastatic breast cancer patients. OncoTargets Ther. 2020, 13, 853–863. [Google Scholar] [CrossRef]
- Yang, Z.; Li, N.; Li, X.; Lei, L.; Wang, X. The prognostic impact of hormonal receptor and HER-2 expression discordance in metastatic breast cancer patients. OncoTargets Ther. 2020, 13, 853–863. [Google Scholar] [CrossRef]