Discussion
In summary, we identified the missense mutation p.V445M of the SCN4A gene from two patients with SCM who were recruited from two individual families. To determine the electrophysiological function of this mutation, a transfected CHO-K1 cell model was employed for the whole-cell patch-clamp recording. Evaluation of the transient Na+ current showed a hyperpolarizing shift in both the activation and inactivation curves of the mutant channel as compared with the WT channel. The window and sustained Na+ currents of the mutant channel were larger than those of the WT. Upon transfection of both Nav1.4 and a Navβ4 peptide, resurgent Na+ currents could be obtained 3~5 ms after a prepulse of depolarization. Compared with that of the WT channel, the magnitude of the resurgent Na+ currents were conspicuously larger in the mutant channel. Moreover, the time to peak of the resurgent Na+ currents in the mutant channel was slower than that of the WT channel, although no difference was found between them in the decay kinetics.
Myotonia results from an involuntary muscle contraction that persists for several seconds after the cessation of voluntary effect [
22]. The hyperexcitability of muscles with a burst of muscle action potentials leads to an after-contraction that can last for several seconds. The spontaneous, painless discharges show a waxing and waning pattern upon EMG study. A sustained burst of discharges can be elicited by a brief voluntary contraction, and the firing frequency is usually 20~80 Hz, which means the interval of the depolarizing potentials is between 12.5 and 50 ms [
23]. The repetitive, high-frequency discharges in myotonia suggest that resurgent Na
+ currents may be a critical factor involving the phenotype. Channels are usually refractory to be activated after inactivation until the membrane potential has been sufficiently hyperpolarized. Nevertheless, channels can reopen, allowing a surge of inward current during repolarization, i.e., resurgent Na
+ currents. Raman and Bean first described resurgent Na
+ currents in cerebellar Purkinje neurons [
16]. In the cerebellum, the resurgent Na
+ currents following relief of an ultra-fast open-channel block by an endogenous blocking particle, probably the C-terminal portion of the Na
vβ4 subunit [
24], may contribute to high-frequency firing [
16,
25]. The Na
v1.6 channel is the major carrier of resurgent Na
+ currents in cerebellar [
26] and DRG [
27] neurons. In the whole-cell configuration of transfected CHO-K1 cells expressing the Na
v1.7 channel, resurgent Na
+ currents after a long depolarizing prepulse have also been identified [
13]. Resurgent Na
+ currents from the mutant Na
v1.7 channel may play an important role in the episodic attacks of severe neuropathic pain found in both erythromelalgia and paroxysmal extreme pain disorders [
28]. Enhancement of resurgent Na
+ currents has even been proposed to be highly correlated with the severity of neuropathic pain [
28]. In the present study, a hyperpolarizing shift was found in both activation and inactivation curves between the mutant and WT channels. Although the window currents increased in the mutant channel, the hyperexcitability with repetitive recurrent discharges could not be fully explained by the changes in the transient Na
+ current alone. Here, we described the resurgent Na
+ currents in the Na
v1.4 channel. Compared with the WT channel, the larger magnitude and slowing of the time to peak of the resurgent Na
+ currents in the mutant channel may be one of the crucial factors of the functional disturbances of SCM. It has been proposed that impaired inactivation is a major determinant of resurgent Na
+ currents in Na
v1.7 channels [
28]. Future research should investigate whether mediation of the inactivation process governs the functional consequences resulting from a mutation in the Na
v1.4 channel, although prolonged recovery of slow inactivation has been observed in the p.V445M mutant channel [
20].
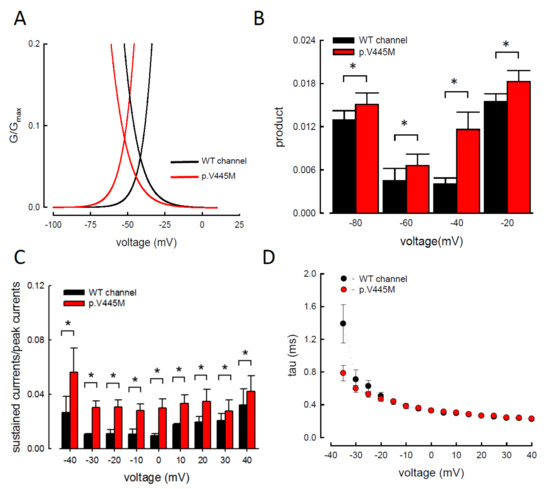
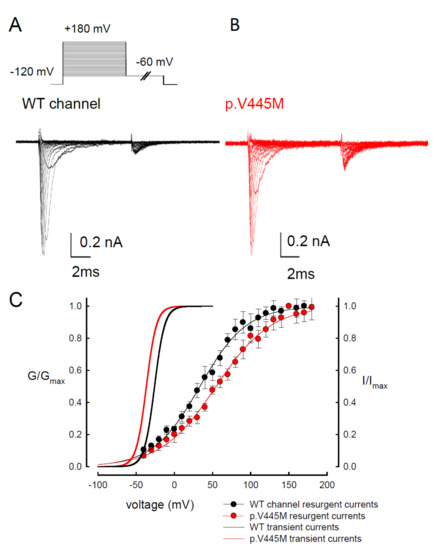
In our study, resurgent currents of Nav1.4 channel are discernible only in the presence of the Na
vβ4 peptides (
Figure S1). According to the prevailing model, this is ascribable to the competition between the Na
vβ4 peptides and the inactivating peptide for the open channel pore [
14,
15,
16,
17]. However, quite other biophysical characteristics, including (1) the concomitant increase of resurgent and sustained currents with the p.V445M mutant channels (A–C and ), (2) the very much different activation curves for the transient and resurgent currents (C) all strongly argue against that the Na
vβ4 peptide competes with the inactivating molecule for the same single open state, (3) the slower time to peak but unchanged decay kinetics of the resurgent currents with p.V445M mutant channels also contradicts that Na
vβ4 peptide competes with the inactivating molecule for the same single open state to generate the resurgent currents. We therefore propose that there may be two distinct open states (with two corresponding inactivated states) of the Na
v1.4 channel (), each responsible for the transient (O
1) and resurgent (O
2) currents, respectively. Significant occupancy of O
2 is possible only in the presence of the Na
vβ4 peptides, which is thus chiefly a gating modifier inducing new gating conformations rather than a pore blocker competing with the inactivating peptides.
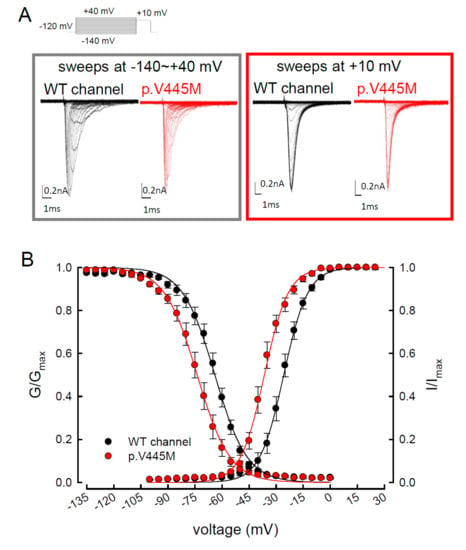
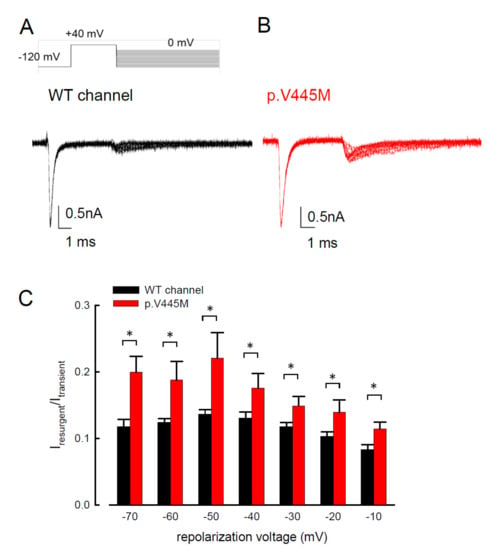
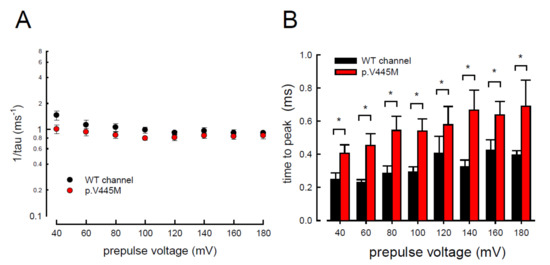
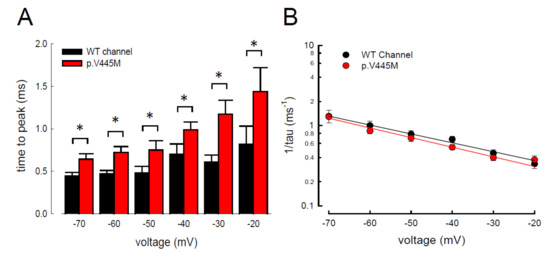
The activation curves of the resurgent Na
+ currents in both the WT and p.V445M mutant channels markedly moved to a more positive voltage range, with a much less steep slope than those of the transient currents (C). We proposed that there may be two open states of the channel and that the gating modification illustrates the electrophysiological changes of the genesis of resurgent Na
+ currents [
13]. This study showed that either after a depolarizing prepulse (B) or under a variable extent of repolarization (A), the time to peak of the resurgent Na
+ currents in the mutant channel was extended significantly compared with that in the WT channel; however, the decay kinetics (1/tau) remained similar between the channels. The p.V445M mutation significantly accelerated the entry and protracted the duration of the new open (activation) state, leading to an increase in the magnitude of the resurgent Na
+ currents. The reopening of the fast inactivated channels was probably accompanied by conformational changes of the domain I/S6 (DI/S6), where V445M is located.
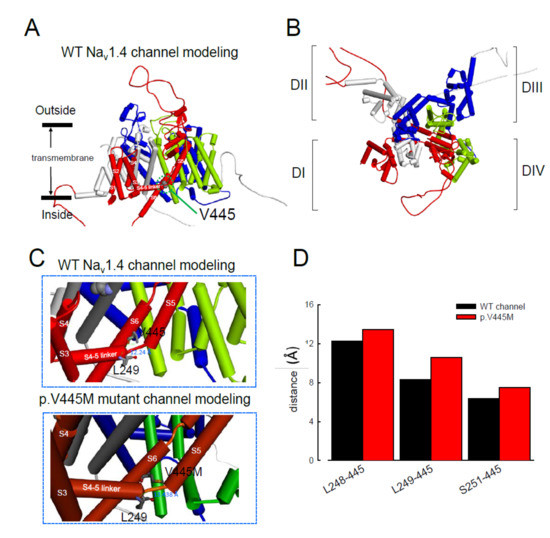
The computer homology model of the WT Na
v1.4 channel evaluated the topological structure change caused by the p.V445M mutation (A,B). The model estimated that the change of Val445 to Met may increase the distance between V445 and the residues at the S4–S5 linker (DI/S4-5 linker) (e.g., L248, L249 and S251; ). While the S4–S5 linker plays an imperative role in governing the activation/inactivation of resurgent Na
+ currents [
13], such a distance change could contribute to the attenuation of inactivation and uncoupling between activation and inactivation. In the previous study, genesis of resurgent Na
+ currents in the Na
v1.7 channel was found to be possibly due to the existence of two open states in activation [
13]. Through modulating the transitional kinetics between these two states, the S4–S5 linker in Domain III seems to play an important role in activation–inactivation coupling. Mutations involving the linker and relevant nearby regions may, therefore, enhance the sustained and resurgent Na
+ currents, leading to marked hyperexcitability and repetitive firing in the corresponding tissues. Herein, we postulated that the change of distance between V445 and the residues of the S4–S5 linker may be one of the critical factors for the coupling effect. Aberrations in modulation of the coupling facilitate the generation of resurgent Na
+ currents and increase both the magnitude and time to peak, augmenting the repetitive firing. The mutation causes slowing of the time to peak of the resurgent Na
+ currents elicited in the Na
v1.4 channel, which increases the firing frequency of the muscle membrane. The action potential propagates to the T-tubule, which induces the opening of the voltage-gated calcium channel from the sarcoplasmic reticulum. The released calcium ion binds to troponin C, which induces the cross bridge to contract. The resurgent Na
+ currents introduce repetitive high-frequency firing, facilitating the binding leading to muscle stiffness. The essential tremors and chronic seizures found in the family members with a p.G1537S mutation in the
SCN4A gene [
29] may also have been attributable to the disturbance of the resurgent Na
+ currents in the Na
v1.4 channel.
Figure 8. Homology modeling of the WT and mutant human Nav1.4 channels. (A) Side view of the homology model shows the transmembrane helixes of the four domains. The four domains I, II, III and IV are colored red, white, blue and green. The side chain of V445 in the S6 linker of Domain I (DI) is indicated in the CPK model. (B) A closer view of the WT Nav1.4 channel from the external side of the pore. (C) A diagram of Domain I of the homology model of the WT Nav1.4 and p.V445M mutant channels is shown in the schematic presentation. The side chains of V445 in the S6 segment and L248, L249 and S251 in the S4–5 linker of Domain I are shown with sticks of various colors. A closer view of the area is shown in the panel, demonstrating inter-residue distances of ~8.3196, ~12.24 and ~6.348 Å between V445 and L248, V445M and L249 and V445 and S251, respectively, in the WT Nav1.4 channel, while the distances between p.V445M and L248, p.V445M and S249 and p.V445M and S251 are ~7.54, ~13.438 and ~10.551 Å, respectively, in the p.V445M mutant channel. (D) The relative distances between the residue V445 and L248, L249 and S251 in the homology modeling is shown. Note the conspicuously decreased distances in all cases in the WT channel compared with those in the p.V445M mutant channel.
In addition to SCM, mutations in Na
v1.4 are also responsible for PMC. Clinically, mutations involving the Arg1448 of the
SCN4A gene are frequently found in patients with PMC [
30]. The resurgent Na
+ currents in the Na
v1.4 channel were first described using rat DRG neurons transfected with Na
v1.4r-R1448P. Jarecki et al. reported that the resurgent Na
+ currents could be obtained and that the average relative amplitude was 4.8± 0.7% [
18], much lower than our findings (~10% in the WT and approximately 15–20% in mutant channels, C). Furthermore, the Arg1448 locates at DIV/S4, which is the outermost extracellular charged residue in the sodium channel voltage sensor that modulates the activation/inactivation coupling of the channel. The study showed that the Na
v1.4r-R1448P channels slow the inactivation and generate resurgent Na
+ currents, suggesting that the mutation slows the rate of open-channel fast inactivation and may be able to induce resurgent Na
+ currents. In another study, large resurgent Na
+ currents could be observed in nearly 62% of Na
v1.8-null DRG neurons using Biolistic transfected with Na
v1.6r during the repolarization stage; however, no resurgent Na
+ currents were identified in neurons transfected with a TTX-R version of the rat skeletal muscle sodium channel Na
v1.4, although they generated a larger peak sodium current amplitude than the Na
v1.6 channels [
27]. The discrepancy may have been due to the different expression systems and the modification of the channel to become TTX-resistant since resurgent Na
+ currents are difficult to obtain using DRG neurons or WT skeletal muscles [
27]. In a recent report, a p.G1306E mutation of SCN4A gene caused NDM and Brugada syndrome after treated with flecainide. The different isoforms of Nav1.4 expressed in the skeletal and cardiac muscles may result in such a rare phenotype [
31].
In conclusion, mutations in SCN4A are responsible for SCM. The hyperpolarizing shifting of the activation and inactivation curves, which results in an increase of window and sustained Na+ currents, may contribute to the hyperexcitability of neural-muscle tissues. The Nav1.4 channel can generate resurgent Na+ currents, which may further increase repetitive firing of the action potential in skeletal muscle, one of the hallmarks of SCM in EMG. In addition to myotonia, patients with SCM frequently experience episodes of muscle weakness. Overall, SCN4A mutations resulting in defects of slow inactivation and delay of time to peak in resurgent Na+ currents are likely factors contributing to constant myotonia and weakness in SCM patients.
- Rosenfeld, J.; Sloan-Brown, K.; George, A.L., Jr. A novel muscle sodium channel mutation causes painful congenital myotonia. Ann. Neurol. 1997, 42, 811–814. [Google Scholar] [CrossRef]
- Takahashi, M.P.; Cannon, S.C. Enhanced slow inactivation by V445M: A sodium channel mutation associated with myotonia. Biophys. J. 1999, 76, 861–868. [Google Scholar] [CrossRef]
- Huang, C.W.; Lai, H.J.; Huang, P.Y.; Lee, M.J.; Kuo, C.C. Anomalous enhancement of resurgent Na(+) currents at high temperatures by SCN9A mutations underlies the episodic heat-enhanced pain in inherited erythromelalgia. Sci. Rep. 2019, 9, 12251. [Google Scholar] [CrossRef] [PubMed]
- Rudel, R.; Lehmann-Horn, F. Membrane changes in cells from myotonia patients. Physiol. Rev. 1985, 65, 310–356. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.M. Differential diagnosis of myotonic disorders. Muscle Nerve 2008, 37, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Grieco, T.M.; Malhotra, J.D.; Chen, C.; Isom, L.L.; Raman, I.M. Open-channel block by the cytoplasmic tail of sodium channel beta4 as a mechanism for resurgent sodium current. Neuron 2005, 45, 233–244. [Google Scholar] [CrossRef]
- Khaliq, Z.M.; Gouwens, N.W.; Raman, I.M. The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: An experimental and modeling study. J. Neurosci. 2003, 23, 4899–4912. [Google Scholar] [CrossRef]
- Afshari, F.S.; Ptak, K.; Khaliq, Z.M.; Grieco, T.M.; Slater, N.T.; McCrimmon, D.R.; Raman, I.M. Resurgent Na currents in four classes of neurons of the cerebellum. J. Neurophysiol. 2004, 92, 2831–2843. [Google Scholar] [CrossRef]
- Cummins, T.R.; Dib-Hajj, S.D.; Herzog, R.I.; Waxman, S.G. Nav1.6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS Lett. 2005, 579, 2166–2170. [Google Scholar] [CrossRef]
- Theile, J.W.; Jarecki, B.W.; Piekarz, A.D.; Cummins, T.R. Nav1.7 mutations associated with paroxysmal extreme pain disorder, but not erythromelalgia, enhance Navbeta4 peptide-mediated resurgent sodium currents. J. Physiol. 2011, 589, 597–608. [Google Scholar] [CrossRef]
- Bergareche, A.; Bednarz, M.; Sanchez, E.; Krebs, C.E.; Ruiz-Martinez, J.; De La Riva, P.; Makarov, V.; Gorostidi, A.; Jurkat-Rott, K.; Marti-Masso, J.F.; et al. SCN4A pore mutation pathogenetically contributes to autosomal dominant essential tremor and may increase susceptibility to epilepsy. Hum. Mol. Genet. 2015, 24, 7111–7120. [Google Scholar] [CrossRef]
- Featherstone, D.E.; Fujimoto, E.; Ruben, P.C. A defect in skeletal muscle sodium channel deactivation exacerbates hyperexcitability in human paramyotonia congenita. J. Physiol. 1998, 506, 627–638. [Google Scholar] [CrossRef]
- Cavalli, M.; Fossati, B.; Vitale, R.; Brigonzi, E.; Ricigliano, V.A.G.; Saraceno, L.; Cardani, R.; Pappone, C.; Meola, G. Flecainide-Induced Brugada Syndrome in a Patient With Skeletal Muscle Sodium Channelopathy: A Case Report With Critical Therapeutical Implications and Review of the Literature. Front. Neurol. 2018, 9, 385. [Google Scholar] [CrossRef] [PubMed]