We live and to do so we must breathe and eat, so are we a combination of what we eat and breathe? Here we will consider this question, and the role in this respect of the AMP-activated protein kinase (AMPK). Emerging evidence suggests that AMPK facilitates central and peripheral reflexes that coordinate breathing and oxygen supply, and contributes to central regulation of feeding and food choice. We propose, therefore, that oxygen supply to the body is aligned with not only the quantity we eat, but also nutrient-based diet selection, and that the cell-specific expression pattern of AMPK subunit isoforms is critical to appropriate system alignment in this respect. If this is the case, then aberrant cell-specific changes in the expression of AMPK subunit isoforms could give rise, in part, to known associations between a wide variety of conditions associated with metabolic disorder.
- AMPK
- breathing
- feeding
- oxygen
- altitude
- hypoxia
- thermogenesis
- mitochondria
- ion channels
- neurons
- pulmonary
- artery
- infection
- diabetes
- obesity
- sleep apnoea
- pulmonary hypertension
1. Introduction
It is evident that we must breathe without serious interruption from birth to death, and we do so with such ease that it almost goes unnoticed, whether we cough, sneeze, snort or, from our partner's perspective, snore. That is, until the onset of certain respiratory diseases, such as sleep-disordered breathing and pulmonary hypertension. These disorders may appear unrelated to each other and to be far removed from food choice and metabolic disease, but evidence of cross-associations is growing.
2. AMPK and the Need to Breathe and Feed
Breathing is coordinated by a sophisticated motor program, which develops in utero in order to coordinate lung ventilation after birth, and responds appropriately to changes in oxygen demand during such activities as exercise, sleep or ascent to altitude. The fundamental rhythmic patterns of ventilatory activity are coordinated by a respiratory central pattern generator (rCPG) through downstream motor outputs from the brainstem and spinal cord, in a similar way to any other type of locomotion or rhythmic behaviour [1]. This occurs independently of peripheral or higher (suprapontine) input, with every breath triggered by cyclical phases of inspiratory muscle contraction (diaphragm, external intercostals), and followed by passive expiration through relaxation of these muscles. In addition, active expiration may be engaged to increase ventilation through recruitment of expiratory muscles (the abdominals and internal intercostals) when the rate of metabolism requires more oxygen. Alternatively, at the end of inspiration, the process of post-inspiration may adapt to reduced requirements by lengthening contraction of the diaphragm and adduction of laryngeal muscles to slow expiratory airflow by increasing airway resistance. These are the principal components of normal breathing (eupnoea), during which the composition and phasic pattern of muscle recruitment/activity is state-dependent. Each component of this cycle is built into the rCPG, central to the activity of which is the pre-Botzinger Complex [2], a brainstem microcircuit from which the rhythm of inspiration originates. In the present context, it is important to note that breathing patterns exhibit acute adaptation when, for example, metabolism increases, and also adapts on longer timescales during growth and maturation, pregnancy, ageing, disease and injury.
Perhaps most important of all is our ability to adapt to deficits in oxygen supply, the most vital elixir of life. This is evident from birth, when hypoxia triggers the first breath due, in part, to the hypoxic ventilatory response (HVR; Figure 1). This response increases ventilatory drive through rCPG, and thus triggers subsequent adaptation to extrauterine life, through maturation of the carotid bodies, our primary peripheral chemoreceptors that drive HVR [3][4][5], and the airways, alveoli and pulmonary vasculature of the lungs that enable optimal gaseous exchange [6]. Gaseous exchange at our lungs is aided by another reflex response to falls in alveolar oxygen availability [7], namely hypoxic pulmonary vasoconstriction (HPV; Figure 1). HPV is a local response mediated by mechanisms intrinsic to the smooth muscles and endothelia of the pulmonary blood vessels, which aids ventilation-perfusion matching, by diverting blood flow through the path of least resistance, from oxygen-deprived to oxygen-rich areas of the lung. By contrast, systemic arteries dilate in response to hypoxia in order to meet the metabolic needs of the tissues they supply [8][9].
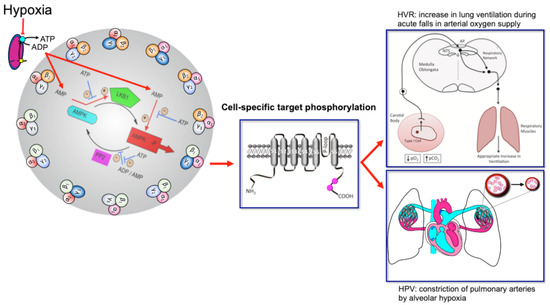
Figure 1. Turn the AMP up to 11 and breathe. Schematic shows the mechanism by which increases in the cellular AMP:ATP and ADP:ATP (adenosine monophosphate and diphosphate to adenosine triphosphate) ratios may activate up to 12 AMPK heterotrimeric subunit combinations, leading to target phosphorylation and induction of the hypoxic ventilatory response (HVR) and hypoxic pulmonary vasoconstriction (HPV). Red arrows indicate pathways leading to AMPK activation by hypoxia, flat heads indicate inhibition, arrow heads indicate activation. Blue arrows with flat heads indicate inhibitory pathways that contribute to the regulation of AMPK activity. Black arrows indicate direction of flow for subordinate pathways. Created by Evans.
The mechanisms of hypoxia-response coupling in those specialised cells that coordinate cardiorespiratory reflex responses to falls in oxygen availability remain keenly debated topics, nowhere more so than with respect to HVR and HPV. However, emerging evidence now strongly suggests that the AMP-activated protein kinase (AMPK) might facilitate both HVR and HPV [10]. This hypothesis [11][12] was built on prior observations that mitochondria of oxygen-sensing cells were exquisitely sensitive to hypoxia, and the proposal that this was due to the selective expression in these cells of a form of cytochrome C oxidase (COX) that was uniquely sensitive to changing PO2 within the physiological range [13][14]. Support for this has been provided not only by the use of mitochondrial inhibitors [15][16][17][18][19][20], but by gene deletion strategies that examined the role of two nuclear encoded atypical subunits of the mitochondrial electron transport chain: (i) NDUFA4L2 encoding NADH dehydrogenase [ubiquinone] 1 alpha subcomplex 4-like 2 (NDUFA4L2) [21]; and (ii) COX4I2 encoding cytochrome c oxidase subunit 4 isoform 2 (COX4I2) [22][23]. NDUFA4L2 is a subunit of complex I, which transfers electrons from NADH to ubiquinone, while COX4I2 is a subunit of cytochrome c oxidase, which catalyses the transfer of electrons from cytochrome c to oxygen. NDUFA4L2 and COX4I2 are constitutively expressed under normoxia not only by oxygen-sensing type I cells of the carotid body [24], but also by pulmonary arterial myocytes [25][26]. In most other cell types NDUFA4L2 and COX4I2 expression is ordinarily low, although their expression may be increased during prolonged hypoxia [22][23]. Accordingly, carotid body type I cell responsiveness to acute hypoxia and acute HVR are abolished in mice by conditional deletion of Cox4I2 in tyrosine hydroxylase expressing catecholaminergic cells [27], while HPV is occluded in isolated, ventilated and perfused lungs from Cox4I2 knockout mice [28]. Therefore, these atypical nuclear encoded subunits not only represent a further distinguishing feature of oxygen-sensing cells, but, at least in the case of COX4I2, appear to be critically important for hypoxia-response coupling within the physiological range of PO2.
Modulation of the properties of COX by COX4I2 is likely critical to hypoxia-response coupling in these highly specialised cells, because COX4I2 lowers the oxygen affinity of COX [29] and removes the inhibition by ATP that is normally conferred by COX4I1 [23][30]. In oxygen-sensing cells, therefore, COX4I2 expression may facilitate rapid decreases in ATP production as oxygen availability falls within the physiological range (~100 mmHg to ~20 mmHg) [31], in part because the rate of mitochondrial oxidative phosphorylation will not increase as ATP levels decline [22][25][30][32]. COX4I2 may thus accelerate increases in adenosine diphosphate (ADP):adenosine triphosphate (ATP) ratios, which would in turn increase adenosine monophosphate (AMP):ATP ratios via the adenylate kinase reaction [33][34], leading ultimately to concomitant activation of AMPK [35][36].
Consistent with this, all agents that inhibit mitochondrial oxidative phosphorylation activate AMPK in an AMP-dependent manner [35][36]. Moreover, there is now a growing realisation that AMPK is capable of phosphorylating targets outside of those canonical pathways by which it regulates cell-autonomous metabolic homeostasis [37][38][39][40][41][42][43][44][45][46]. In this way, it appears that, during evolution, the role of AMPK in regulating metabolic homeostasis has been extended through natural selection to support system-level control of substrate supply (e.g., of oxygen, glucose and fatty acids) in order to maintain energy (ATP) homeostasis across the whole body [11].
This entry is adapted from the peer-reviewed paper 10.3390/ijms21103518
References
- Smith, J.C.; Abdala, A.P.; Borgmann, A.; Rybak, I.A.; Paton, J.F.; Brainstem respiratory networks: Building blocks and microcircuits. Trends Neurosci. 2013, 36, 152–162, .
- Yang, C.F.; Feldman, J.L.; Efferent projections of excitatory and inhibitory prebotzinger complex neurons. J. Comp. Neurol. 2018, 526, 1389–1402, .
- Kumar, P.; Prabhakar, N.R. Peripheral chemoreceptors: Function and plasticity of the carotid body. Compr. Physiol. 2012, 2, 141–219.
- Nurse, C.A. Synaptic and paracrine mechanisms at carotid body arterial chemoreceptors. J. Physiol. 2014, 592, 3419–3426.
- Zera, T.; Moraes, D.J.A.; Da Silva, M.P.; Fisher, J.P.; Paton, J.F.R. The logic of carotid body connectivity to the brain. Physiology 2019, 34, 264–282.
- Demosthenes Papamatheakis; Arlin B. Blood; Joon H. Kim; Sean M. Wilson; Antenatal hypoxia and pulmonary vascular function and remodeling.. Current Vascular Pharmacology 2013, 11, 616-640, 10.2174/1570161111311050006.
- U. S. V. Euler; G. Liljestrand; Observations on the Pulmonary Arterial Blood Pressure in the Cat. Acta Physiologica Scandinavica 1946, 12, 301-320, 10.1111/j.1748-1716.1946.tb00389.x.
- Sylvester, J.T.; Shimoda, L.A.; Aaronson, P.I.; Ward, J.P. Hypoxic pulmonary vasoconstriction. Physiol. Rev. 2012, 92, 367–520.
- Roy, C.S.; Sherrington, C.S. On the regulation of the blood-supply of the brain. J. Physiol. 1890, 11, 85–185.
- A. Mark Evans; AMPK breathing and oxygen supply. Respiratory Physiology & Neurobiology 2019, 265, 112-120, 10.1016/j.resp.2018.08.011.
- Evans, A.M. Amp-activated protein kinase and the regulation of ca2+ signalling in o2-sensing cells. J. Physiol. 2006, 574, 113–123.
- Evans, A.M. Hypoxia, cell metabolism, and cadpr accumulation; Springer: Boston, MA, USA, 2004.
- Mills, E.; Jobsis, F.F. Simultaneous measurement of cytochrome a3 reduction and chemoreceptor afferent activity in the carotid body. Nature 1970, 225, 1147–1149.
- Mills, E.; Jobsis, F.F. Mitochondrial respiratory chain of carotid body and chemoreceptor response to changes in oxygen tension. J. Neurophysiol. 1972, 35, 405–428.
- Firth, A.L.; Yuill, K.H.; Smirnov, S.V. Mitochondria-dependent regulation of kv currents in rat pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L61–L70.
- Post, J.M.; Hume, J.R.; Archer, S.L.; Weir, E.K. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am. J. Physiol. 1992, 262, C882–C890.
- Duchen, M.R.; Biscoe, T.J. Relative mitochondrial membrane potential and [ca2+]i in type i cells isolated from the rabbit carotid body. J. Physiol. 1992, 450, 33–61.
- Duchen, M.R.; Biscoe, T.J. Mitochondrial function in type i cells isolated from rabbit arterial chemoreceptors. J. Physiol. 1992, 450, 13–31.
- Buckler, K.J.; Turner, P.J. Oxygen sensitivity of mitochondrial function in rat arterial chemoreceptor cells. J. Physiol. 2013, 591, 3549–3563.
- Wyatt, C.N.; Buckler, K.J. The effect of mitochondrial inhibitors on membrane currents in isolated neonatal rat carotid body type i cells. J. Physiol. 2004, 556, 175–191.
- Daniel Tello; Eduardo Balsa; Barbara Acosta-Iborra; Esther Fuertes-Yebra; Ainara Elorza; Angel Ordoñez; María Corral-Escariz; Inés Soro; Julián Aragonés; Ester Perales-Clemente; et al. Induction of the Mitochondrial NDUFA4L2 Protein by HIF-1α Decreases Oxygen Consumption by Inhibiting Complex I Activity. Cell Metabolism 2011, 14, 768-779, 10.1016/j.cmet.2011.10.008.
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. Hif-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122.
- Huttemann, M.; Kadenbach, B.; Grossman, L.I. Mammalian subunit iv isoforms of cytochrome c oxidase. Gene 2001, 267, 111–123.
- Ting Zhou; Ming‐Shan Chien; Safa Kaleem; Hiroaki Matsunami; Single cell transcriptome analysis of mouse carotid body glomus cells. The Journal of Physiology 2016, 594, 4225-4251, 10.1113/jp271936.
- Aras, S.; Pak, O.; Sommer, N.; Finley, R., Jr.; Huttemann, M.; Weissmann, N.; Grossman, L.I. Oxygen-dependent expression of cytochrome c oxidase subunit 4-2 gene expression is mediated by transcription factors rbpj, cxxc5 and chchd2. Nucleic Acids Res. 2013, 41, 2255–2266.
- Huttemann, M.; Lee, I.; Gao, X.; Pecina, P.; Pecinova, A.; Liu, J.; Aras, S.; Sommer, N.; Sanderson, T.H.; Tost, M.; et al. Cytochrome c oxidase subunit 4 isoform 2-knockout mice show reduced enzyme activity, airway hyporeactivity, and lung pathology. FASEB J. 2012, 26, 3916–3930.
- Moreno-Dominguez, A.; Ortega-Saenz, P.; Gao, L.; Colinas, O.; Garcia-Flores, P.; Bonilla-Henao, V.; Aragones, J.; Huttemann, M.; Grossman, L.I.; Weissmann, N.; et al. Acute o2 sensing through hif2alpha-dependent expression of atypical cytochrome oxidase subunits in arterial chemoreceptors. Sci. Signal. 2020, 13, eaay9452, .
- Natascha Sommer; Maik Hüttemann; Oleg Pak; Susan Scheibe; Fenja Knoepp; Christopher Sinkler; Monika Malczyk; Mareike Gierhardt; Azadeh Esfandiary; Simone Kraut; et al. Mitochondrial Complex IV Subunit 4 Isoform 2 Is Essential for Acute Pulmonary Oxygen Sensing. Circulation Research 2017, 121, 424-438, 10.1161/CIRCRESAHA.116.310482.
- David Pajuelo Reguera; Kristýna Čunátová; Marek Vrbacký; Alena Pecinová; Josef Houštěk; Tomáš Mráček; Petr Pecina; Cytochrome c Oxidase Subunit 4 Isoform Exchange Results in Modulation of Oxygen Affinity. Cells 2020, 9, 443, 10.3390/cells9020443.
- Susann Horvat; Cordian Beyer; Susanne Arnold; Effect of hypoxia on the transcription pattern of subunit isoforms and the kinetics of cytochrome coxidase in cortical astrocytes and cerebellar neurons. Journal of Neurochemistry 2006, 99, 937-951, 10.1111/j.1471-4159.2006.04134.x.
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C.; Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253, .
- K. M. Kocha; K. Reilly; D. S. M. Porplycia; J. McDonald; T. Snider; Christopher D. Moyes; Evolution of the oxygen sensitivity of cytochrome c oxidase subunit 4.. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 2014, 308, R305-R320, 10.1152/ajpregu.00281.2014.
- Dzeja, P.P.; Terzic, A. Phosphotransfer networks and cellular energetics. J. Exp. Biol. 2003, 206, 2039–2047.
- Panayiotou, C.; Solaroli, N.; Karlsson, A. The many isoforms of human adenylate kinases. Int. J. Biochem. Cell Biol. 2014, 49, 75–83.
- Auciello, F.R.; Ross, F.A.; Ikematsu, N.; Hardie, D.G. Oxidative stress activates ampk in cultured cells primarily by increasing cellular amp and/or adp. FEBS Lett. 2014, 588, 3361–3366.
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of ampk activation. Cell Metab. 2010, 11, 554–565.
- Andersen, M.N.; Skibsbye, L.; Tang, C.; Petersen, F.; MacAulay, N.; Rasmussen, H.B.; Jespersen, T. Pkc and ampk regulation of kv1.5 potassium channels. Channels 2015, 9, 121–128.
- Mia, S.; Munoz, C.; Pakladok, T.; Siraskar, G.; Voelkl, J.; Alesutan, I.; Lang, F. Downregulation of kv1.5 k channels by the amp-activated protein kinase. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2012, 30, 1039–1050.
- Moral-Sanz, J.; Mahmoud, A.D.; Ross, F.A.; Eldstrom, J.; Fedida, D.; Hardie, D.G.; Evans, A.M. Amp-activated protein kinase inhibits kv 1.5 channel currents of pulmonary arterial myocytes in response to hypoxia and inhibition of mitochondrial oxidative phosphorylation. J. Physiol. 2016, 594, 4901–4915.
- Lipton, A.J.; Johnson, M.A.; Macdonald, T.; Lieberman, M.W.; Gozal, D.; Gaston, B. S-nitrosothiols signal the ventilatory response to hypoxia. Nature 2001, 413, 171–174.
- Murphy, B.A.; Fakira, K.A.; Song, Z.; Beuve, A.; Routh, V.H. Amp-activated protein kinase and nitric oxide regulate the glucose sensitivity of ventromedial hypothalamic glucose-inhibited neurons. Am. J. Physiol. Cell Physiol. 2009, 297, C750–C758.
- Zhang, J.; Dong, J.; Martin, M.; He, M.; Gongol, B.; Marin, T.L.; Chen, L.; Shi, X.; Yin, Y.; Shang, F.; et al. Ampk phosphorylation of ace2 in endothelium mitigates pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 509–520.
- Ahmadi, M.; Roy, R. Ampk acts as a molecular trigger to coordinate glutamatergic signals and adaptive behaviours during acute starvation. eLife 2016, 5, e16349.
- Schneider, H.; Schubert, K.M.; Blodow, S.; Kreutz, C.P.; Erdogmus, S.; Wiedenmann, M.; Qiu, J.; Fey, T.; Ruth, P.; Lubomirov, L.T.; et al. Ampk dilates resistance arteries via activation of serca and bkca channels in smooth muscle. Hypertension 2015, 66, 108–116.
- Ross, F.A.; Rafferty, J.N.; Dallas, M.L.; Ogunbayo, O.; Ikematsu, N.; McClafferty, H.; Tian, L.; Widmer, H.; Rowe, I.C.; Wyatt, C.N.; et al. Selective expression in carotid body type i cells of a single splice variant of the large conductance calcium- and voltage-activated potassium channel confers regulation by amp-activated protein kinase. J. Biol. Chem. 2011, 286, 11929–11936.
- Ikematsu, N.; Dallas, M.L.; Ross, F.A.; Lewis, R.W.; Rafferty, J.N.; David, J.A.; Suman, R.; Peers, C.; Hardie, D.G.; Evans, A.M. Phosphorylation of the voltage-gated potassium channel kv2.1 by amp-activated protein kinase regulates membrane excitability. Proc. Natl. Acad. Sci. USA 2011, 108, 18132–18137.