miRNAs role in hormone signaling pathways in CRC drug resistance and their potential as future targets for overcoming resistance to treatment.
- microRNA,hormone signaling
1. The Role of Hormone Signaling in CRC Resistance
In receiving cells, hormone signaling comprises hormone binding to its receptor and signal transduction through different functional pathways. Several hormone signaling pathways are altered in CRC, often resulting in drug resistance. Tumor cells can avoid drug-mediated apoptosis at different levels of signaling pathways as these typically have multiple components. Most hormone signaling pathways exert their function through PI3K/Akt cascade: Upon hormone stimulation, PI3K is phosphorylated and its activity is mediated by RasandSrcfamilykinases that phosphorylate Akt, which, in turn, regulates the activation of proteins involved in apoptosis, metabolism, protein synthesis, and cell division [44]. Furthermore, the 3R3 subunit of PI3K is upregulated in CRC and controls the expression of TP via the NF-kB pathway, thus preventing the activation of drugs (5-FU, capecitabine) and resulting in chemoresistance [37]. PI3K/mTOR inhibition in CRC patient-derived cells has also been reported to reinstate cetuximab resistance [45]. PI3K action is regulated by the tumor suppressor phosphatase and tensin homolog (PTEN). Altered pathways responding to insulin, estrogen, and androgen involve PTEN [46], resulting in cetuximab and panitumumab resistance [47]. Thus, PI3K/Akt/mTOR signaling is effectively involved in many hormonal pathway-related resistances, though it is not the only one. Upon binding of ligands (such as insulin and growth factors) to their tyrosine kinase receptor [48], PTEN has also been involved in cetuximab resistance by altering Ras pathway activity [49]. MAPK/ERK pathway was found downstream of many growth factor receptors often overexpressed in CRC [50], such as insulin-like growth factor receptor (IGFR) [51]. MAPKs are involved in several cellular functions such as proliferation, differentiation, and apoptosis through three primary components: p38, ERK, and JNK. Hyperactivation of p38 was recently implicated in oxaliplatin resistance in CRC [52]. MAPKs exert their functions through NF-kB [46], activated in 60–80% of CRC patients [53], and involved in conferring chemoresistance through inhibition of apoptosis [54]. In other studies, CRC cells resistant to 5-FU showed high expression of TCF4 and β-catenin, indicating an altered Wnt pathway [55]. Since hormone signaling is triggered upon binding of the hormone to its receptor, expression of hormone receptors in recipient cells is important in modulating the impact of this interaction, as well as influencing downstream cascades.
1.1. Insulin/Insulin-Like Growth Factor Signaling Pathway in CRC
The insulin/IGF system plays a crucial role in CRC development [56]. Insulin/IGF bind to the insulin receptor (IR), which is converted into its activated phosphorylated form. High levels of pIR were observed in the transition phase from normal epithelium to adenocarcinoma, indicating the key role of insulin/IR in CRC progression [57]. IR substrates (IRS)-1 and -2 also contribute to disease development: IRS-1 overexpression is associated with metastasis [58], whereas IRS-2 is related to early-stage tumor formation. IRS-2 overexpression increases PI3K pathway activity, leading to Akt phosphorylation and reduced cell adhesion in colon cancer [59]. IGF-I and IGF-II are significantly overexpressed in CRC [60]. High IGF serum levels were detected in CRC patients and found to act through autocrine/paracrine as well as endocrine signaling [51]. IGF regulates cell survival, proliferation, and metabolism by binding to IGFR or IR. IGF-1R was found overexpressed in CRC [61] where it resulted in Akt activation and upregulation of the anti-apoptotic protein Bcl-xL [62]. IGF-1R is associated with drug resistance as it regulates the expression of multidrug-resistance-associated protein 2 (MRP2). For this purpose, Shen et al. silenced IGF-1R which subsequently drove the nuclear translocation of nuclear-factor-like2 and blockage of MRP2 gene expression [63]. Low MRP2 expression causes an increase in intracellular drug concentration and accumulation, suggesting how the IGF-1R signaling pathway may foster drug efflux and tumor cell survival despite therapy. Further analysis of these hormone receptors revealed that silencing of IGF-1R increases the sensitivity of CRC cells to 5 -FU, mitomycin C, oxaliplatin, and vincristine [63].
1.2. Thyroid Hormone Signaling Pathway in CRC
Animal studies and clinical data from CRC patients suggest that thyroid status may impact tumor formation, growth, metastasis, and drug sensitivity [64,65,66]. This physiological hormonal system comprises both triiodothyronine (T3) and thyroxine (T4) hormones. T4 is the primary inactive thyroid product, which is converted into active T3 by type 1 or 2 deiodinase enzymes (D1 and D2, respectively). In contrast, type 3 deiodinase (D3) transforms T4 and T3 into inactive metabolites [65,67]. Deiodinases represent the link between thyroid hormone signaling and β-catenin pathway. The β-catenin/TCF complex regulates D3 expression at the transcriptional level resulting in high expression of D3 in CRC. Indeed, using E-cadherin to sequester β-catenin, a reduction in D3 expression occurs.Low D3 levels lead to increased expression of T3, resulting in inhibited proliferation of CRC cells [68]. Furthermore, T3 may induce resistance to oxaliplatin, 5-FU, and their combination (FOX) by downregulating Akt/PI3K in CRC stem cells [69]. Basically, T3 binds to its receptors (TRα and TRβ), regulates the expression of target genes via thyroid hormone receptor response elements [70]. Thyroid hormone receptor (TR) β1 expression is downregulated in CRC, prompting cancer cells to proliferate and migrate via PI3K/Akt signaling modulation. Overexpression of TRβ1 induces a reduction of inactivated Akt, disrupting its pathway and suppressing CRC cell progression and migration [71]. In contrast, TRα1 overexpression and Wnt signaling pathway upregulation promote tumor formation in CRC [72] by contributing to escape from chemotherapy-induced apoptosis [73]. Indeed, further evidence report that T3 affects drug resistance targeting BMP4/Wnt pathway [70].
1.3. Additional Altered Hormone Signaling in Resistant CRC Cells
Other hormonal systems contribute to drug resistance in CRC cells. Leptin is an adipokine involved in regulating hyperphagia, glucose homeostasis, growth, immune response, and angiogenesis [74]. Leptin and its receptors (ObR) are highly expressed in CRC [75], indeed, leptin inhibition reduces tumor growth [76] and leads to an overexpression of ObR, which inhibits 5-FU-induced apoptosis [77]. Moreover, high levels of growth hormone (GH) were identified in CRC patients, where the hormone enhances cell proliferation, survival, and oncogenicity through autocrine signaling [78]. GH also plays a role in the acquisition of drug-induced apoptosis resistance by inhibiting the expression of PPARγ and BAX in CRC cells, allowing evasion from therapy-mediated DNA damage [79]. Corticotropin-releasing hormone (CRH) is a peptide hormone involved in stress response that exerts its function by binding to its specific receptors, including corticotropin-releasing hormone receptor2 (CRHR2), which exhibits a significantly reduced expression in CRC. Fas is one of the CRHR2/Ucn2 signaling targets in CRC and alternation in Fas expression prevents Fas-mediated apoptosis, thus conferring resistance to therapy [80]. The lower incidence of CRC in women than in men highlights the importance of estrogen as a biological protective effector in gastrointestinal disease [81,82]. Estrogens can bind to estrogen receptor α (ERα) orβ (ERβ). In normal colon regions, ERβ is implicated in epithelium maintenance and growth, and in immune system modulation, while decreased ERβ expression promotes the risk of CRC partly by influencing gut permeability [83]. Reduced ERβ expression in CRC was also correlated with increased proliferation and inhibition of apoptosis [84]. ERα downregulation in CRC [85] suggests that this receptor might contribute to drug resistance via regulatory effect in the expression of pro- or anti-apoptotic proteins, such as cyclin D1 [84]. Estrogen signaling might be indirectly involved in conferring resistance in CRC cells since a deficiency in ER signaling can be compensated for by other signaling pathways in these cells [86,87]. Estrogen-related receptor alpha (ERRα) is also involved in the same system. ERRα is an orphan nuclear receptor since its endogenous ligand has not yet been identified. Interestingly, it is constitutively active even without any ligand binding [88] and regulates the expression of many genes, most of which are also targeted by ERα [89]. ERRα is overexpressed in CRC [83] and its upregulation is responsible for resistance to trametinib [90], making ERRα a potential new target in CRC.
2. Role of miRNA in Hormone Signaling in Therapy-Resistant CRC
miRNAs are known as posttranscriptional regulators of gene expression, affecting about 60% of overall protein-coding genes [91], thereby modulating developmental, physiological, and pathological processes. Recent reports described their critical role in the regulation of hormone signaling, including expression modulation of hormone receptor-coding genes and genes related to hormonal intracellular signaling cascades [92,93,94]. Moreover, miRNAs can be modulated by hormones [95], thus suggesting a reciprocal flux which anyway revises cancer cell sensitivity to treatment. Several deregulated miRNAs are associated with drug resistance in CRC (Table 1), many of which are directly involved in hormone signaling. The following sections unfold the involvement of miRNAs into hormonal signaling pathways whose interactions are responsible for decreased therapy success by overcoming drug-mediated apoptosis and, thereby, inducing survival in CRC cells.
Table 1. Drug resistance-associated miRNAs in CRC.
| miRNA | Expression | Targets | Resistance | Reference |
|---|---|---|---|---|
| miR-21 | ↑ | PDCD4 TIAM1 PTEN TGFBR2 CDC25A |
5-FU | [120] |
| miR-143 | ↓ | NF-kB Bcl-2; IGF-1R |
5-FU | [94,121] |
| miR-365; miR-224 | ↓ | - | 5-FU | [122] |
| miR-34a | ↓ | SIRT1 | 5-FU | [123] |
| miR-494 | ↓ | DPYD | 5-FU | [124] |
| miR-302 | ↓ | IGF-1R | 5-FU | [133] |
| miR-10b | ↑ | BIM | 5-FU | [134] |
| miR-375-3p | ↓ | TYMS | 5-FU | [135] |
| miR-20a | ↑ | BNIP2 | 5-FU OxaliplatinTeniposide |
[136] |
| miR-320 | ↓ | FOXM1 | 5-FU Oxaliplatin |
[127] |
| miR-34a | ↑ | - | Oxaliplatin | [129] |
| let-7g | ↓ | Cyclin D Myc E2F |
S-1 | [125] |
2.1. miRNAs and Insulin-Like Growth Factor (IGF) Receptor
IGFs are implicated in tumorigenesis of different cancers, such as prostate, breast, and CRC [51]. As explained in sub-paragraph 2, both IGFs and IGF-R were found overexpressed in CRC, and such boosted IGF-I/IGF-IR signaling pathway enhances cell survival and resistance to chemotherapy [62], a process that apparently involves CIMP. The chromosomal segment 17p13.1 is frequently deleted in CRC, resulting in reduced expression of both miR-497 and miR-195, as their encoding sequences are located in this chromosomal region [96]. Guo et al. proposed miR-497 to have a role in inhibiting IGF-IR signaling pathway, indeed, in CRC, low miR-497 is associated with higher IGF-IR levels, which, in turn, modulates cell death. Restoration of miR-497 expression could be useful for inhibition of IGF-IR in CRC and resistance to apoptosis by blocking overactivation of survival signaling pathways [96]. Many other miRNAs are implicated in IGFR signaling modulation resulting in drug resistance. As a matter of fact, miR-143 targets IGF-1R resulting in oxaliplatin resistance [97] and, indeed, overexpression of miR-143 was reported to re-sensitize CRC cells to such therapeutic by caspases activation [97]. Interestingly, Xu et al. found that miR-143 is downregulated in blood as well, suggesting its potential as a prognostic biomarker in CRC. Besides miR-143, IGF-1R is also a target of miR-302, which downregulation in CRC may cause 5-FU resistance. Liu et al., actually found that miR-302 overexpression reduces 5-FU resistance as demonstrated by enhanced cell death, subsequently, therapeutic treatment. This re-sensitizing action is due to the direct repression of IGF-1R associated with consequent inhibition of Akt, frequently activated in chemo-resistant CRC cells [98]. Lastly, a further miRNA is able to modulate IGFRs expression skewing CRC therapeutic response. In this respect, miR-185 was found to be involved in radio-resistance in CRC cells by targeting IGF-1R and IGF-2R. In detail, miR-185 is downregulated in colon cancer, leading to increased expression of its targets and, thus, resulting in refractory to ionizing radiation, as it was confirmed by improved CRC cell radio-sensitivity upon restoration of the miRNA expression [99].
2.2. miRNAs and IGF-R Downstream Components
Besides receptors, miRNAs impair hormone signaling also at the downstream level, resulting in ectopic pathways and then in drug resistance. Among such regulators, miR-1260b targets PDCD4, one of the effectors of the PI3K/Akt signaling pathway reported to regulate apoptosis [100] and the response to drug treatment [101]. miR-1260b expression was reported increased in CRC patients and linked to 5-FU therapy resistance. Accordingly, the use of a miR1260b inhibitor confirmed the involvement of such miRNA in 5-FU-resistance in CRC, further revealing to be mediated by inhibition of PI3K/Akt signaling pathway. Interestingly, the overexpression of IGF-I in CRC enhanced the activation of such pathway, indicating that miR-1260b may regulate drug sensitivity via IGF signal modulation [102]. IRS-1 is reported to be an oncogene involved in the regulation of angiogenesis, metastasis, and even chemosensitivity, thereby, IRS-1 increased levels due to the reduction of its regulatory miRNA expression may be responsible for lowering the therapy responsiveness in CRC. In the same manner, miR-145 exhibits a tumor suppressor activity in CRC by modulating Myc [103], STAT1 [104] and other genes including IRS-1 [105].In fact, miR-145 downregulation was shown to increase tumor survival [106], a feature sharply in contrast with the therapeutic success. Lastly amidst other miRNAs, miR-128 is downregulated in CRC, failing to restrain IRS-1 and resulting in inhibited apoptosis [107].
2.3. miR-155 and Adrenaline
Previous examples have exhibited a definite trend, where miRNAs were responsible for the refractory phenotype in CRC by skewing hormone signaling pathways. Nevertheless, there is evidence that a different course exists since hormones may affect miRNAs expression. The miR-155/adrenaline coupling is one example, as adrenaline was found to increase miR-155 expression, leading to drug resistance [108]. CRC cells significantly express adrenergic receptors [109] whose ligands are catecholamines, including adrenaline. Adrenaline was previously shown to induce 5-FU resistance in CRC in vitro by upregulating ABCB1 transporter [109].Indeed, mouse cancer models exposed to stressful stimuli to set up catecholamine signals, exhibited restrained response to anti-tumor therapy [110]. In this regard, adrenaline showed to activate NF-kB signaling in CRC cells, increasing the expression of miR-155, which, in turn, favored cell proliferation and reduced sensitivity to cisplatin [110], corroborating the potential of catecholamines in endowing resistance to CRC.
2.4. miR-7 and Corticotropin-Releasing Hormone Receptor
Nowadays, immune therapy has emerged as a rising approach to the conventional anti-tumor treatments since cancer cells were shown to express a large amount of inhibitory molecules, such as PD-L1 [111], CD80/CD86 [112], which undermine the immunological response through binding immune checkpoint receptors such as PD-1 and CTLA4 [113]. Another way exploited by cancer cells to deceive the immune-mediated cytotoxicity is the escape from Fas/FasL-mediated apoptosis [114,115,116]. Although cancer treatments, such as radiotherapy, exhibited a successful increase in Fas expression [115], immune escape is still a current challenge and the CRH system seems to be involved in it. Rodriguez et al. reported that CRHR2 expression is reduced in CRC cells supporting tumor survival, proliferation, EMT, metastasis, and resistance [117]. In vitro and in vivo studies showed that miR-7 expression is induced by CRHR2 signaling and its ectopic expression enhances apoptosis and cell cycle arrested by negatively regulating YY1 expression [118]. Thus, YY1 expression benefits from reduced CRHR signaling and its overexpression impairs Fas expression. Collectively, CRHR2/Ucn2 signaling sustains CRC cell resistance to Fas/FasL-mediated apoptosis by targeting miR-7/YY1/Fas circuit [80], as it was confirmed by increased CD-11 (Fas agonist)-mediated apoptosis after restoration of CRHR2 signaling.
3. Novel Therapeutic Agents in the Treatment of Drug Resistance in CRC
Many therapeutics acting on hormone signaling have been tested towards resistant CRC. In CRC, neurokinin receptor 1 (NK1R) antagonists reduced tumor growth through inhibition of Wnt/β-catenin and Akt/mTOR signaling pathways. The NKIR antagonist also inhibited cancer stem cells, which are thought to confer higher resistance to therapy [119]. Besides molecule antagonists, hormone analogs have been tested in CRC. Lee et al. exploited a T4 analog (named Tetrac) to block CRC progression competing with the endogenous hormone to bind and, thus, block the surface integrin receptor αvβ3. Its usage, in combination with cetuximab, was reported to inhibit cell proliferation and CRC xenografts [120]. Brigatinib, which inhibits IGF-1R [121], and Amiodarone, a Thyroid Hormone Receptor antagonist [122], were studied with encouraging results. Significant outcomes have also been obtained with natural products in resistant CRC cells. Gambogic acid is a natural compound extracted from the Garciniahanburyi plant [123]. It acts by interfering with MAPK signaling via JNK pathway, which, in turn, induces apoptosis and inhibits proliferation of 5-FU-resistant CRC cells [123]. The China Food and Drug Administration has approved its use in phase II clinical trials for the treatment of solid tumors. Piperlongumine, a natural constituent of the fruit of the long pepper (Piper longum), inhibits PI3K and Ras, reducing the activity of Akt/NF-kB, c-Myc, cyclin D1, blocking CRC cell growth, proliferation and survival. It also induces mitochondrial apoptosis, downregulating Bcl-2 [124]. In addition to the molecules that directly modulate pathway component activity and considering the emerging role of miRNAs in regulating the expression of genes involved in hormone signaling pathways, recent studies have been focused on strategies to regulate oncomiR expression, including antisense anti-miR oligonucleotides, locked nucleic acid anti-miRs, miRNA sponges and small molecule inhibitors of miRNAs (SMIRs) [125]. ASMIR against miR-21, named AC1MMYR2, was developed by Shi et al., who studied the three-dimensional structure of the Dicer binding site on pre-miR-21 to find a molecule able to block miR-21 maturation. AC1MMYR2 inhibits the expression of miR-21 and consequently increases the expression of its targets: PTEN (Figure 1), PDCD4 and RECK [126]. This axis is involved in 5-FU resistance (Table 1). miR-1260b inhibitor was tested by Zhao et al., who showed the reduction of PDCD4 expression and p-Ant and p-PI3K levels in IGF-R signaling pathway, thus, re-sensitizing CRC to 5-FU treatment [102]. One of the most used anti-miRs in CRC is anti-miR-135b [127,128]. Upregulation of miR-135b in CRC reduces apoptosis and increases cell growth by regulating the expression of TGF-βR2, DAPK1, APC and FIH by activating APC/β-catenin and Src-PI3K pathways. Anti-miR-135b was therefore used in a CRC mouse model, resulting in reduced proliferation and increased apoptosis [129] and drug sensitivity [128]. SD-208, a TGF-β receptor kinase inhibitor, downregulated miR-135b expression [127]. Among other targets, miR-135b affects FOXO1 and ERRα expression, which is associated with apoptosis modulation (Figure 2). FOXO1 is also involved in IR signaling cascade [130]. Thus, pre-treatment with SD-208 improved chemosensitivity in CRC cells resistant to 5-FU [127]. Melatonin was shown to exert a robust anti-tumor effect through modulation of cell cycle dynamics and apoptosis [131]. Recently, Sakatani et al. investigated the molecular mechanism of melatonin in 5-FU-resistant CRC cells [132], suggesting that melatonin strongly promotes apoptosis in 5-FU-resistant CRC cells by blocking thymidylate synthase (TYMS) activity, pointing to an inverse correlation between TYMS expression levels and 5-FU sensitivity in CRC. TYMS is one of the main downstream targets of miR-215-5p. Melatonin upregulates miR-125-5p expression, enhancing the suppressive effect of miR-125-5p on TYMS expression in 5-FU-resistant cells, thus, leading to inhibition of proliferation and increased apoptosis in these resistant cells [132].
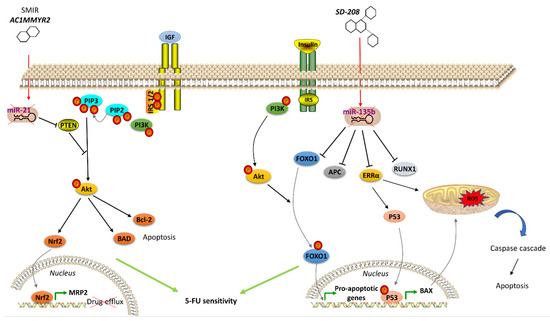
Figure 1. Agents acting on hormone signaling to revert drug resistance in CRC. Left, the small molecular inhibitor of specific miRNA (SMIR) AC1MMYR2 represses miR-21, which regulates PTEN expression, thus modulating IGFR signaling pathway and resulting in inhibited drug efflux and induced apoptosis in resistant CRC cells. Right, SD-208 inhibits miR-135b, which controls expression of proteins (including FOXO1 in IR signaling pathway) involved in apoptosis regulation, thus resulting in apoptosis of resistant CRC cells. (↑ = miRNA overexpression) (↓ = miRNA downexpression)
This entry is adapted from the peer-reviewed paper 10.3390/cells10010039