Inflammatory bowel disease (IBD) represents an umbrella term for the chronic remission and relapse of immunologically-mediated idiopathic diseases. IBD is generally diagnosed under two major classifications as Crohn’s disease (CD) and ulcerative colitis (UC) with significantly contrasting etiologies. Multiple studies over the decades have still remarkably left the pathogenesis of the diseases an unresolved mystery. CD tends to occur in any part of the gastrointestinal tract (GIT) and is associated with complications, whereas UC, on the other hand, is strictly restricted to the inflammation of the colon. The onset of the diseases is marked at young adulthood in genetically susceptible individuals responding to commensal microbes or environmental cues like poor hygiene, unbalanced dietary intake, a lack of physical exercise, smoking, and stress.
- inflammatory bowel disease (IBD)
- Crohn’s disease (CD)
- ulcerative colitis (UC)
1. Conventional Therapies for IBD
The application of common therapeutics depends on the severity of the disease and the highly affected areas (Figure 1). Conventional treatment for IBD includes aminosalicylates (ASAs), systemic corticosteroids, topical corticosteroids, antibiotics, immunomodulators, and biologic therapies. ASAs for a very long time have been modified by linking 5-ASA (therapeutic moiety) through an azo bond with sulfa pyridine (carrier) to form sulfasalazine to treat IBD. ASAs are broken down in the ileocolic tract by colonic bacteria [1], boosting immunity against pathogenic bacterial antigens and also reducing inflammation by inhibiting NF-κB, IL-1, and IL-2 [2]. Unfortunately, 5-ASA and its various new preparations [3] such as olsalazine, mesalazine, and balsalazide have a poor bioavailability with various hematological side effects in addition to dose-dependent diarrhea, nausea, vomiting, abdominal pain, and fatigue [4]. Traditionally, patients with extensive mild-to-moderate active UC are given oral corticosteroids as the first line of therapy in case they fail to respond to topical mesalazine [5]. Glucocorticosteroids have also been used for the treatment of IBD, and 80% of patients have shown positive treatment responses [6]. However, the toxicity of the drugs has been linked to infections related to Candida spp. with an increase in blood glucose, thus compromising the glucometabolic balance in non-diabetic individuals [7]. The long term usage of corticosteroids can lead to diabetes, Cushing’s syndrome [8], and osteoporosis [9]. Budesonide and beclomethasone dipropionate (BDP) are used as topical glucocorticoids that are absorbed through the mucosa into the bloodstream and inactivated by the liver [10]. Budesonide is the first line of treatment for mildly active CD [11]. However, is not as effective as a standard glucocorticoid capable of stimulating remission in CD and also do not prevent CD relapse [9]. Budesonide was found to be less effective than systemic steroid course for inducing remission in active CD. Budesonide was also found to not be effective in the prevention of relapsing after medically or surgically-induced remission [12]. The adverse effects of budesonide are acne, weight gain, mood swings, moon face, and hair loss [13].
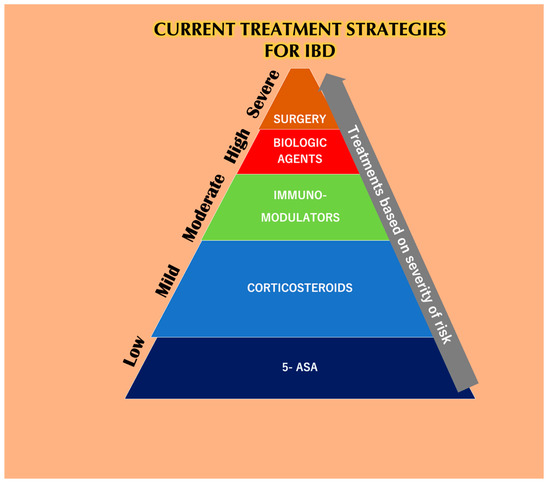
Figure 4. The current treatment strategies for IBD depends solely upon the severity of the disease. At lower stages of IBD, 5-aminosalicylic acid (5-ASA) helps reduce the symptoms, but not for a prolonged period because it has a reduced bioavailability. Corticosteroids are also prescribed to IBD patients in the initial stages but are burdened with systemic toxicities. Immuno-modulators are used during moderate cases of IBD but have been reported to be linked with various side effects and also lead to the risk of developing lymphoma. Biological agents like anti-TNF-α agents are used when patients are least responsive to the previous methodologies. Unfortunately, these agents are also responsible for various side effects when taken alone or as a combination with other mentioned therapies. Finally, IBD patients associated with dysplasia or cancer who undergo surgery also have a chance of relapsing [14].
Antibiotics like ciprofloxacin, metronidazole, and rifaximin used for the treatment for IBD and controlling bacterial overgrowth are considered safe and well-tolerated [15]. Nonetheless, the continued treatment with ciprofloxacin can induce insomnia, acute psychosis, convulsions, dizziness, and mild-to-severe phototoxicity [16]. The adverse effects of metronidazole include anorexia, nausea, dizziness, encephalopathy, diarrhea, seizure, cerebellar ataxia, and peripheral neuropathy [17]. Common symptoms of rifaximin are nausea, abdominal pain, flatulence, vomiting, and urticarial skin reactions when consumed in high doses [18]. Thiopurines generally focus on maintenance therapy and are linked with severe side effects in more than 30% of IBD patients, along with drug discontinuation in more than 20–40% of IBD patients [19]. Thiopurines are mostly recommended for patients with steroid-dependent UC. Patients who have experienced early or frequent relapses while taking mesalazine or are intolerant depend on thiopurines. Studies have demonstrated that thiopurines can be given for at least five years in UC [5]; the most common side effects are pancreatitis, vomiting, nausea, cutaneous eruption, hepatitis [20], and a higher risk for lymphoma [21]. Methotrexate, on the other hand, is only effective in the activation and continuance of remission in CD but not in the case of UC [22], and it can induce hepatic toxicity, bone marrow suppression, gastrointestinal intolerance, and hypersensitive pneumonitis [23]. Thiopurines and methotrexate do not induce remission in severe CD, but they are considered for long term maintenance. Methotrexate is also embryotoxic and contraindicated during pregnancy because it can cause anencephaly, hydrocephaly, and meningomyelocele, thus limiting its use in young women [24]. When applied intravenously, cyclosporine A has been found to be effective in patients with severe, steroid-refractory UC [25]. However, cyclosporine A causes dose-dependent adverse effects like renal toxicity, lymphoma, hypertension, microbial infections (staphylococcus sepsis), seizures, and anaphylaxis [25].
Biological therapies including monoclonal antibodies against TNF-α and α4 integrins have been developed in the last 15 years to facilitate the treatment of IBD. Infliximab was one of the first biologicals to be approved by the food and drug administration (FDA) for the therapy of severe, active, and fistulizing CD [26]. Infliximab can bind with mouse mTNF-α and thus induce the lysis of mTNF-α-expressing cells through antibody-dependent cellular cytotoxicity in vitro. Infliximab can also induce apoptosis in the monocytes and lamina propria lymphocytes via caspase-8, -9, and -3 in patients with CD by specifically binding to mTNF-α [27]. Studies have established that single-dose infliximab is safe and effective in the management of acute CD. Patients receiving maintenance infliximab that was used as a replacement for single-dose infliximab seemed to reduce the use of corticosteroids. Patients receiving concurrent immunosuppressive therapy developed a low incidence of antibodies against infliximab. However, the development of malignancy is linked with infliximab therapy [28]. Infusion reactions to infliximab offer multiple signs and symptoms within two hours of administration, and some of these reactions can be life-threatening. Additionally, the combination therapy of infliximab with azathioprine and methotrexate does not seem to confer an advantage over infliximab monotherapy [29]. Subsequently, the further development of anti-TNF-α antibodies like adalimumab and golimumab concerning the treatment for IBD have come into action [30] A recent study conducted on pediatric patients with CD showed that the weekly dosing of adalimumab was clinically beneficial in children who experienced nonresponse or flare on every other week dosing. Out of the 83 patients who escalated to blinded weekly dosing, 24.1% achieved remission and 51.8% achieved response. The highest rate of remission occurred those who escalated to 40 mg weekly. Abdominal and anal abscesses, as well as device-related sepsis, were observed as serious infections in patients whose doses were escalated [31]. Adalimumab and golimumab cause mild-to-moderate injection site reactions [32]. Swapping anti-TNF-α for another drug is frequently practiced when a patient becomes unreactive to one agent due to intolerance and secondary or primary failure [33]. Bone marrow toxicity (neutropenia, thrombocytopenia, and anemia) can also occur during anti-TNF-α treatment [34]. When combined with thiopurines, anti TNF-α agents were reported to cause a risk of lymphoma in patients, particularly those with CD [35]. An increased risk of opportunistic infection risk has also been associated with anti-TNF-α agents in CD patients [36]. Vedolizumab is a new immunomodulator that is effective in moderate-to-severe IBD, but it has been associated with infections, infusion-related reactions, and malignancies [37].
1.1. Probiotics
The complex interactions of diet, normal intestinal microbiota, and health have encouraged the introduction of probiotics that exert beneficial effects on the host [38]. Many microbiotas have been examined for relieving intestinal dysbiosis and bacteria like Lactobacillus, Bifidobacterium, and Streptococcus have been selected in the formulation of probiotics because they have shown a clinical effect on gastrointestinal inflammation and the ability to maintain normal human intestinal microbiota [39]. Probiotics have been used to understand the efficacy of living microorganisms in alleviating the symptoms of IBD [40]. VSL#3, a probiotic mixed with four strains of Lactobacilli (Lactobacilli case, Lactobacilli acidophilus, Lactobacilli delbrueckii sub spp., and Bulgaris), three strains of Bifidobacterium (Bifidobacterium longum, Bifidobacterium breve, and Bifidobacterium infantis), and a Streptococcus strain (Streptococcusalivarius sub spp. thermophilus) have been shown to be efficient in yielding remission in mild-to-moderately active UC [41]. One or more genetically modified strains have also been found to be more beneficial. For example, in a phase I clinical trial of treating Crohn’s disease using Lactococcus lactis strain, the thymidylate synthase gene exchanged using synthetic sequence encoding mature human IL-10 showed a decrease in disease activity and avoided systemic side effects [42]. Additionally, clinical trials of probiotics for the treatment of IBD are very infrequent. In an experiment conducted on 20 human volunteers (13 men and 7 women) who had a history of IBD, colon cancer, a recent antibiotic or anti-coagulant therapy was supplemented with dietary fibers and probiotics separately and in combination; the authors found no significant differences in the fecal SCFA concentrations, and no significant effect was found on epithelial proliferation [43]. E. coli Nissle 1917 and Saccharomyces boulardi probiotics did not show any significant effect in the remission of IBD and had no advantage compared with placebos [44]. Therefore, more intense understanding is required while selecting a probiotic strain when there is a disruption in the intestinal environment by disease, diet, and antibiotics that can, in turn, affect the health of the host.
1.2. Fecal Microbiota Transplantation
Fecal microbiota transplantation (FMT) involves the exchange of intestinal microbiota from healthy donors to re-establish intestinal microbiota in diseased individuals. FMT has been clinically adapted to recurrent Clostridium difficile, which leads to loss of microbiota diversity and the expansion of facultative anaerobic bacteria [45]. For the treatment of Clostridium difficile, FMT showed high rates of cure, regardless of the donor, recipient, and delivery method. However, some patients develop repeated infections and permanent changes in their gut microbiota upon FMT treatment for Clostridium difficile [46]. Clinical studies focusing on the efficacy and safety of FMT in IBD have multiplied over the years, but various factors like the selection of microbiota, immune response, and environmental factors are to be considered important factors in the pathogenesis of IBD [47]. The remission rate of FMT for patients with UC has been reported as 33%, but the long-term durability and safety are still a concern. However, there a previous study found no significant improvement in patients with CD following FMT [48]. Therefore, well-designed studies are required before randomized clinical trials in IBD [49].
2. Nano Drug Delivery Systems as an Alternative
The application of nanotechnology in biomedical research arena has advanced by leaps and bounds over the past few decades, especially for cancer therapies and regenerative medicine. The unprecedented success in providing a safer alternative treatment option to current conventional cancer therapies is commendable. The extrapolation of the biomedical applications of nanotechnology or nano-drug delivery systems for the treatment of IBD is still in its infancy, though accumulating evidence has made it look promising. Since the current course of treatment for IBD-related disorders mostly comprises anti-inflammatory agents, corticosteroids, immunosuppressants, and biologic agents, these agents mainly contribute to maintaining remission from inflammatory actions, thus additionally complicating the patient profile by contributing adverse side effects. Henceforth, the discovery of alternative methods of the local delivery of these conventional agents to the inflamed tissue provides a rationale to incorporate nano-drug delivery systems in the treatment of IBD. Moreover, assistance from the use of natural compounds and conventional therapies delivered via nano-drug delivery systems could help to maintain the remission and relapse of the disease, thus improving therapeutic efficacies and avoiding systemic side-effects.
Nano-drug delivery passively or actively targets the site of inflammation and has been proven to be more beneficial than conventional therapies. Due to nano-drug delivery’s structured morphology, effective targeting, increased bioavailability, and requirement of a low concentration of the drug in unhealthy tissue, minimized, systemic adverse effects are highly anticipated [50]. The reduced size of the nanoparticles (1–1000 nm) facilitates the improved and careful transport of active molecules to the inflamed tissue via the epithelial enhanced permeability and retention effect (EPR) and promotes the selective uptake of nanoparticles by the immune cells at the target site [50]. The paracellular transport of the carrier with the drug is made permeable when the intestinal epithelial barrier is compromised [51]. The transcellular transport (transcytosis) of the nanoparticles begins with endocytosis at the cell apical membrane, followed by the release of the nanoparticles at the basolateral pole, where they come in contact with the immune cells present at the submucosal layer. The physicochemical properties of the nanoparticles (NPs), the physiology of the GIT, and the animal model are used to determine the NPs’ proper intake [52].
2.1. Surface Charge-Dependent Drug Delivery Systems
The surface charge of nanoparticles can be modified to influence the electrostatic interactions with the components of the GIT. Due to the presence of sulfates and sialic acid residues, colonic mucins tend to carry a negative charge. An excessively increased mucus production can be observed in CD, and this provokes a thick mucus layer in the affected area [53]. Positively charged nanoparticles can easily adhere to the negative mucosal surface within the inflamed tissues due to electrostatic interaction and thus promote cellular uptake and drug release through the better contact with the mucosal surface [54]. It was found that cationic polymethacrylate (Eudragit RL) nanoparticles (120 nm diameter) loaded with clodronate enables a complete drug release that (compared to the free clodronate) significantly decreases the myeloperoxidase activity (MPO) in the 2,4,6-trinitrobenzene sulfonic acid (TNBS) and oxazolone (OXA)-induced colitis through ionic interactions with the dissolution medium or mucin [55]. However, cationic nanoparticles have been found to have adhered to the mucosal layer and become immobilized, thus leading to the premature release of the drug, probably due to the presence of a negative mucosal surface and strong electrostatic adhesion. This was seen in the case of chitosan functionalized poly(lactic-co-glycolic acid) (PLGA) nanoparticles targeted ex vivo to intestinal mucosa adhered to the mucosal surface with minimal translocation and accumulation in both healthy and inflamed mucosa [56]. In contrast to positively charged proteins like eosinophil cationic protein and transferrin, the development of anionic nanoparticles can tackle these issues in inflamed tissues [57]. They can promote electrostatic repulsion with the negatively charged mucus, thus enabling the anionic nanoparticles to interdiffuse in the mucus network without any interactions, thus alleviating concerns regarding immobilization like in the case of cationic nanoparticles [58]. The negatively charged nanoparticles can target the inflamed mucosa and gradually release the drug depending on the microenvironment of the inflamed intestine [59]. An ex vivo study conducted on neutral, positively charged, and negatively charged liposomes to target colitis induced by dinitrobenzene sulfonic acid (DNBS) found that the adherence of anionic liposomes to inflamed colonic mucosa was two-fold more than neutral or cationic liposomes. This adherence was dependent on the negative charge on the liposomes due to the presence of 12,dimyristoyl-sn-glycerol-3-(phosphor-rac-(1-glycerol)) (DSPG), whereas cationic and neutral liposomes did not significantly bind to the inflamed intestinal mucosa [60]. Though anionic NPs are found to be specific in drug delivery, additional approaches are necessary to improve bioavailability in the colon.
2.2. ROS-Responsive Delivery System
Slight damage to antioxidant defense systems can lead to oxidative stress and cause an abnormal rise in the release of ROS by inflammatory cells like neutrophils and macrophages. Tackling ROS-mediated oxidative stress has been a focus for pharmaceutical strategies to improve targeted drug delivery in diseased colonic sites. Biopsies of patients with UC have found increases in mucosal ROS concentrations up to 10- to 100-fold, and redox-responsive nano-delivery systems have thus become associated with the treatment of UC [61]. Wilson et al. [62] synthesized thioketal nanoparticles formulated from polymer poly (1,4-phenylacetone dimethylene thioketal) that degrade in response to ROS for target delivery of TNF-α small interfering ribonucleic acids (siRNA)complexed with cationic species such as 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP). DOTAP enhances stability in transfection, mucosal transport, and internalization inside the cell, as well as endosomal escapes to intestinal inflammation sites, in mice. Thioketal NPs diminish TNF-α messenger ribonucleic acids (mRNA) levels and protected UC. In another study, to treat colitis in mice, the oral delivery of low molecular weight TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl) using nitroxide radical-containing nanoparticles (RNPsO), made up of an amphiphilic block copolymer and methoxy-poly(ethylene glycol)-b-poly(4-(2,26,6-tetramethyl-piperidine-1-oxyl)oxymethylstyrene(MeO-PEG-b-PMOT), demonstrated that the stable nitroxide radicals on the hydrophobic segment of this copolymer can successfully scavenge ROS [63]. Interestingly, RNPsO were further studied to examine their effect on colonic microflora during UC, and it was found that commensal bacteria like E. coli and Staphylococcus sp. led to a remarkably high dextran sodium sulfate (DSS)-induced colitis in mice, whereas the oral administration of RNPsO outstandingly reduced these commensal bacteria [64]. However, several hindrances, like the faster release of the drug and the instability of these nanocarriers in the lower pH and enzyme-rich environments of the upper GI tract, limit the application of ROS-responsive systems.
2.3. pH-Dependent Drug Delivery System
The pH-sensitivity of nanoparticles as a pharmaceutical strategy enables them to retain and protect their cargo, but are very likely to dissolve or swell in higher pH environments like in the colon, thus allowing for drug release [65]. When a drug directly encounters variations in pH, it becomes redundant in its activity due to extreme oxidation, deamination, or hydrolysis [66]. A synthetic polymer is often coated with pH-dependent coating polymers like methacrylic acid co-polymers (Eudragit®) for oral delivery [67]. Liposomes coated with Eudragit® S100 display appropriate pH response release characteristics when the polymer retains the liposomal release of the drug at pH levels of 1.4 and 6.3—resembling the stomach and small intestine, respectively—but release the drug similar to plain liposomes NPs at a pH of 7.8 (ileocecal junction). However, in vivo conditions, due to the additional challenges of bile salts that cause the premature degradation of liposomes, can result in the early release of the drug in the duodenum [68]. The instability of liposomes in the GIT has pushed researchers to focus on polymer-based nanocarriers for supreme colon-specific drug delivery. Tacrolimus-loaded PLGA NPs encapsulated inside Eudragit® P4135F microspheres showed zero drug or NP release at a pH of 4, but at a pH of 7.4, the NPs were released undamaged [69]. The formulation was found to be only moderately effective, and the overall poor performance was associated with the fabrication of the delivery system where the outer microspheres and the inner NPs had the same solubilities in the organic solvents. Therefore, it was difficult to check on NP integrity while being coated by the microspheres, and there are also chances for the lipophilic drug to be redeposited from the NPs into the microspheres, thus reducing the effect of the drug with partial deposition at the target site [69][70][71]. Hence, the mixing of PLGA and Eudragit® was found to be more effective. The formulation of pH-sensitive NPs using PLGA and Eudragit® loaded with budesonide (BSD) showed a sustained release of the drug at the colonic pH, along with more therapeutic effects than BSD alone when used to treat an TNBS-induced animal model of colitis [72]. Beloqui et al. [67] prepared nanoparticles using pH-sensitive PLGA and Eudragit® S100 loaded with the anti-inflammatory agent curcumin, which is preferential in accumulating at the inflamed region in in vitro and in vivo studies. The permeation of the drugs was found to be enhanced by curcumin-loaded NPs across Caco-2 monolayers and also reduced TNF-α secretion through LPS-activated macrophages (J774 cells) in comparison to the curcumin suspension. In in vivo conditions, NPs were found to significantly reduce neutrophil infiltration while retaining a colonic structure identical to the control group in a murine DSS-induced colitis model. Ribeiro et al. [73] fabricated a drug delivery system by coating pectin on chitosan/layered double hydroxide biohybrid beads loaded with 5-ASA for protection against degradation at the upper GIT. Coating with pectin was able to navigate through the gastric juices and promote the release of the drug from the bio-nano composite beads due to swelling of pectin at pH 7.4. Cyclosporine loaded in PLGA coated with Eudragit® S100 nanospheres were able to generate a sustained release at a pH of 7.4, thus suggesting its capability in UC therapy [74]. Additionally, pH-sensitive Eudragit® S100/ethylcellulose nanofibers loaded with budesonide showed the supreme release of the drug at a pH of 7.4, which was similar to that of spherical NPs [75]. In another in vitro analysis, 5-ASA-loaded chitosan NPs coated with Eudragit® S100 revealed that the drug release was only at the pH values of the colon [76]. Chitosan and alginate coated with Eudragit® S100 pH-sensitive microcrystals also illustrated pH-dependent dexamethasone release, avoiding drug release in the acidic pH conditions of the stomach and small intestine. This enabled the release of the drug in the colonic pH and alleviated inflammation in a DSS-induced mouse colitis model [77]. Though pH-dependent NPs have shown tremendous results in preclinical studies, the variability of pH in IBD patients’ colons shows that a colonic drug delivery system based only on GIT pH would not be reliable [78].
2.4. Biodegradable Drug Delivery Systems
When developing a drug delivery system, the chief goal is to protect the hydrophobic therapeutically active molecules prematurely subjected to degradation for enhancing sustained release at the targeted site and to avoid causing an undesired side effect. One of the ideal methods would be controlled-release systems that could maintain the drug concentration and frequency of administration [79]. Polysaccharides like pectin, chitosan, and alginate have been studied for the oral delivery of hydrophobic drugs for targeting inflammation in the colon [80]. High-water content hydrogel is a cross-linked polymer network that provides physical similarity to biological tissues and thus has an exceptional biocompatibility. Hydrogels can encapsulate hydrophilic drugs with minimal denaturation and aggregation upon exposure to organic solvents [81]. Laroui et al. [82] developed a hydrogel using chitosan and alginate that was cross-linked using Ca2+ and SO2- to encapsulate polylactic acid (PLA) NPs with the anti-inflammatory tripeptide Lys–Pro–Val (KPV). Upon reaching the inflamed colon, the hydrogel was degraded and successfully reduced colitis symptoms, MPO activity, and histologic alterations in a DSS colitis model. In vivo studies have indicated that microparticles made of resistant starch such as high amylose cornstarch loaded with 5-ASA have a high tolerance against the acidic and enzymatic conditions of the upper GIT and can accurately release the drug in the colon [83]. A nanoparticle-in-microparticle oral delivery system (NiMOS) was also designed for colon targeting by encapsulating plasmid and siRNA in type B gelatin nanoparticles. These nanoparticles were loaded in poly(epsilon-caprolactone) (PCL) microspheres that can withstand protein/enzymatic degradation in the upper GIT. The NP release occurs at the inflamed sites of the intestine due to the action of the lipase enzyme on PCL present at the location [84]. Pectin-based microspheres/nanospheres resonate a viable oral colon-specific drug carrier since the gut bacteria like Bacteroides thetaiotamicron and E. coli are capable of degrading the pectin in the colon [85]. Crosslinked pectin microspheres loaded with indomethacin in vitro showed an increased delivery at pH 7.4 compared to non-crosslinked microspheres. Similarly, Eudragit-coated pectin microspheres were also found to be excellent in the colon-specific delivery of the drug [86]. In a recent study, mesalazine-loaded calcium pectin–silica gel beads were developed to control the release of mesalazine in the colon. These beads showed a reduced delivery of mesalazine in a simulated upper GIT condition due to decreased swelling that, in turn, improved the strength of the bead. An elated drug level was found in the simulated colonic fluid with an increased sensitivity of pectin towards the pectinase [87]. In another study, Eudragit® FS30D-coated alginate microspheres filled in hydroxypropyl methylcellulose (HMPC) capsules ensured the release in the colon even though Eudragit FS30D had a solubility before reaching a pH of 7. In vitro studies in simulated colonic fluid with rat fecal content confirmed the bacterial degradability of the alginate, thus prematurely hindering the drug release in the upper GIT. In vivo studies have also shown a marked reduction in the ulcer index in rats treated with microspheres [88]. Resveratrol is a naturally therapeutic agent, but it is also a hydrophobic drug, and the necessity of a hydrophilic carrier is therefore of utmost importance. Biocompatible and non-toxic poly(2-hydroxyethyl methacrylate) and a pH-sensitive poly(N,N-dimethylaminoethyl methacrylate) loaded with resveratrol were integrated into a chitosan matrix gel. The drug was released in a sustained release pattern due to the presence of the chitosan network, thus proposing a versatile tool that can bestow therapeutic benefits in the treatment of IBD [89]. An acetic acid-induced colitis rabbit model was used to study the effect of the quercetin drug, a natural polyphenol in a chitosan-based colon targeted delivery system to selectively target the inflamed colon. The drug-loaded microparticles were more therapeutically effective than a plain drug [90].
Edible plant-derived nanoparticles have also been designed for a novel and nontoxic delivery system to target colon tissues, thereby reducing IBD-mediated inflammation [88]. Zhang et al. [91] developed grapefruit-derived nanovesicles loaded with methotrexate that are biodegradable, biocompatible, and stable across a wide range of pH conditions. The methotrexate nanovesicles were able to downregulate IL-1β and TNF-α by upregulating the release of heme oxygenase-1 (HO-1) in intestinal macrophages and had improved anti-inflammatory properties against DSS-induced IBD compared to a free drug. The oral administration of ginger-derived nanoparticles was also found to increase IEC proliferation and elevate the concentration of anti-inflammatory cytokines by reducing the concentration of proinflammatory cytokines like TNF-α, IL-1β, and IL-6 [92].
This entry is adapted from the peer-reviewed paper 10.3390/nano10122460
References
- Khan, A.K.A.; Piris, J.; Truelove, S.C. An Experiment to Determine the Active Therapeutic Moiety of Sulphasalazine. Lancet 1977, 310, 892–895.
- Zhou, E.M.Z.S.Y.; Fleisher, D.; Pao, L.H.; Li, C.; Winward, B. Intestinal Metabolism and Transport of 5-aminosalicylate. Drug Metab. Dispos. 1999, 27, 479–485.
- Head, K.A.; Jurenka, J.S. Inflammatory bowel disease part I: Ulcerative colitis—Pathophysiology and conventional and alternative treatment options. Altern. Med. Rev. 2003, 8, 247–283.
- Feagan, B.G.; MacDonald, J.K. Oral 5-aminosalicylic acid for induction of remission in ulcerative colitis. In Cochrane Database of Systematic Reviews; Feagan, B.G., Ed. John Wiley & Sons, Ltd.: Chichester, UK, 2012.
- Gionchetti, P.; Rizzello, F.; Annese, V.; Armuzzi, A.; Biancone, L.; Castiglione, F.; Comberlato, M.; Cottone, M.; Danese, S.; Daperno, M.; et al. Use of corticosteroids and immunosuppressive drugs in inflammatory bowel disease: Clinical practice guidelines of the Italian Group for the Study of Inflammatory Bowel Disease. Dig. Liver Dis. 2017, 49, 604–617.
- Dignass, A.; Lindsay, J.O.; Sturm, A.; Windsor, A.; Colombel, J.; Allez, M.; D'Haens, G.; D'Hoore, A.; Mantzaris, G.; Novacek, G.; et al. Second European evidence-based consensus on the diagnosis and management of ulcerative colitis Part 2: Current management. J. Crohn’s Colitis 2012, 6, 991–1030.
- Toruner, M.; Loftus Jr, E.V.; Harmsen, W.S.; Zinsmeister, A.R.; Orenstein, R.; Sandborn, W.J.; Colombel, J.F.; Egan, L.J. Risk Factors for Opportunistic Infections in Patients With Inflammatory Bowel Disease. Gastroenterology 2008, 134, 929–936.
- Peppa, M.; Boutati, E.; Krania, M.; Raptis, S. Hypertension and other morbidities with Cushing's amprsquos syndrome associated with corticosteroids: A review. Integr. Blood Press. Control. 2011, 4, 7.
- Ford, A.C.; Bernstein, C.N.; Khan, K.J.; Abreu, M.T.; Marshall, J.K.; Talley, N.J.; Moayyedi, P. Glucocorticosteroid Therapy in Inflammatory Bowel Disease: Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2011, 106, 590–599.
- Fasci Spurio, F.; Aratari, A.; Margagnoni, G.; Clemente, V.; Moretti, A.; Papi, C. Low bioavailability and traditional systemic steroids in IBD: Can the former take over the latter? J. Gastrointest. Liver Dis. 2013, 22.
- Rutgeerts, P.; Lofberg, R.; Malchow, H.; Lamers, C.; Olaison, G.; Jewell, D.; Danielsson, A.; Goebell, H.; Thomsen, O.O.; Lorenz-Meyer, H.; et al. A Comparison of Budesonide with Prednisolone for Active Crohn’s Disease. N. Engl. J. Med. 1994, 331, 842–845.
- Papi, C.; Luchetti, R.; Gili, L.; Montanti, S.; Koch, M.; Capurso, L. Budesonide in the treatment of Crohn’s disease: A meta-analysis. Aliment. Pharmacol. Ther. 2000, 14, 1419–1428.
- Schoon, E.J.; Bollani, S.; Mills, P.R.; Israeli, E.; Felsenberg, D.; Ljunghall, S.; Persson, T.; Hapten-White, L.; Graffner, H.; Porro, G.B.; et al. Bone mineral density in relation to efficacy and side effects of budesonide and prednisolone in Crohn’s disease. Clin. Gastroenterol. Hepatol. 2005, 3, 113–121.
- Al-Sukhni, W.; McLeod, R. S.; MacRae, H.; OʼConnor, B.; Huang, H.; and Cohen, Z. Oncologic Outcome in Patients With Ulcerative Colitis Associated With Dyplasia or Cancer Who Underwent Stapled or Handsewn Ileal Pouch-Anal Anastomosis. Dis. Colon Rectum. 2010, 53, 1495–1500.
- Gionchetti, P.; Dignass, A.; Danese, S.; Magro Dias, F.J.; Rogler, G.; Lakatos, P.L.; Adamina, M.; Ardizzone, S.; Buskens, C.J.; Sebastian, S.; et al. 3rd European Evidence-based Consensus on the Diagnosis and Management of Crohn’s Disease 2016: Part 2: Surgical Management and Special Situations. J. Crohn’s Colitis 2017, 11, 135–149.
- Tomé, A.M.; Filipe, A. Quinolones. Drug Saf. 2011, 34, 465–488.
- Kuriyama, A.; Jackson, J.L.; Doi, A.; Kamiya, T. Metronidazole-Induced Central Nervous System Toxicity. Clin. Neuropharmacol. 2011, 34, 241–247.
- Scarpignato, C.; Pelosini, I. Rifaximin, a Poorly Absorbed Antibiotic: Pharmacology and Clinical Potential. Chemotherapy 2005, 51, 36–66.
- Chaparro, M.; Ordas, I.; Cabre, E.; Garcia-Sanchez, V.; Bastida, G.; Penalva, M.; Gomollon, F.; Garcia-Planella, E.; Merino, O.; Gutierrez, A.; et al. Safety of Thiopurine Therapy in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2013, 19, 1404–1410.
- Saibeni, S.; Virgilio, T.; D’Inca, R.; Spina, L.; Bortoli, A.; Paccagnella, M.; Peli, M.; Sablich, R.; Meucci, G.; Colombo, E.; et al. The use of thiopurines for the treatment of inflammatory bowel diseases in clinical practice. Dig. Liver Dis. 2008, 40, 814–820.
- Vos, A.C.W.; Bakkal, N.; Minnee, R.C.; Casparie, M.K.; de Jong, D.J.; Dijkstra, G.; Stokkers, P.; van Bodegraven, A.A.; Pierik, M.; van der Woude, C.J.; et al. Risk of malignant lymphoma in patients with inflammatory bowel diseases: A Dutch nationwide study. Inflamm. Bowel Dis. 2011, 17, 1837–1845.
- Engel, M.A.; Neurath, M.F. New pathophysiological insights and modern treatment of IBD. J. Gastroenterol. 2010, 45, 571–583.
- Feagan, B.G.; Fedorak, R.N.; Irvine, E.j.; Wild, G.; Sutherland, L.; Steinhart, A.H.; Greenberg, G.R.; Koval, J.; Wong, C.J.; Hopkins, M.; et al. A Comparison of Methotrexate with Placebo for the Maintenance of Remission in Crohn’s Disease. N. Engl. J. Med. 2000, 342, 1627–1632.
- Lichtenstein, G.R.; Abreu, M.T.; Cohen, R.; Tremaine, W. American Gastroenterological Association Institute Technical Review on Corticosteroids, Immunomodulators, and Infliximab in Inflammatory Bowel Disease. Gastroenterology 2006, 130, 940–987.
- Naganuma, M.; Fujii, T.; Watanabe, M. The use of traditional and newer calcineurin inhibitors in inflammatory bowel disease. J. Gastroenterol. 2011, 46, 129–137.
- Dretzke, J.; Edlin, R.; Round, J.; Connock, M.; Hulme, C.; Czeczot, J.; Fry-Smith, A.; McCabe, C.; Meads, C. A systematic review and economic evaluation of the use of tumour necrosis factor-alpha (TNF-α) inhibitors, adalimumab and infliximab, for Crohn’s disease. Health Technol. Assess. (Rockv.) 2011, 15.
- Danese, S. Mechanisms of action of infliximab in inflammatory bowel disease: An anti-inflammatory multitasker. Dig. Liver Dis. 2008, 40, S225–S228.
- Hanauer, S.B.; Feagan, B.G.; Lichtenstein, G.R.; Mayer, L.F.; Schreiber, S.; Colombel, J.F.; Rachmilewitz, D.; Wolf, D.C.; Olson, A.; Bao, W.; et al. Maintenance infliximab for Crohn’s disease: The ACCENT I randomised trial. Lancet 2002, 359, 1541–1549.
- Lichtenstein, G.R.; Diamond, R.H.; Wagner, C.L.; Fasanmade, A.A.; Olson, A.D.; Marano, C.W.; Johanns, J.; Lang, Y.; Sandborn, W.J. Clinical trial: Benefits and risks of immunomodulators and maintenance infliximab for IBD-subgroup analyses across four randomized trials. Aliment. Pharmacol. Ther. 2009, 30, 210–226.
- Sandborn, W.J.; Feagan, B.G.; Marano, C.; Zhang, H.; Strauss, R.; Johanns, J.; Adedokun, O.J.; Guzzo, C.; Colombel, J.; Reinisch, W.; et al. Subcutaneous Golimumab Induces Clinical Response and Remission in Patients With Moderate-to-Severe Ulcerative Colitis. Gastroenterology 2014, 146, 85–95.
- Dubinsky, M.C.; Rosh, J.; Faubion Jr, W.A.; Kierkus, J.; Ruemmele, F.; Hyams, J.S.; Eichner, S.; Li, Y.; Huang, B.; Mostafa, N.M.; et al. Efficacy and Safety of Escalation of Adalimumab Therapy to Weekly Dosing in Pediatric Patients with Crohnʼs Disease. Inflamm. Bowel Dis. 2016, 22, 886–893.
- Sandborn, W.J.; Hanauer, S.B.; Rutgeerts, P.; Fedorak, R.N.; Lukas, M.; MacIntosh, D.G.; Panaccione, R.; Wolf, D.; Kent, J.D.; Bittle, B.; et al. Adalimumab for maintenance treatment of Crohn’s disease: Results of the CLASSIC II trial. Gut 2007, 56, 1232–1239.
- Gisbert, J.P.; Marín, A.C.; McNicholl, A.G.; Chaparro, M. Systematic review with meta-analysis: The efficacy of a second anti-TNF in patients with inflammatory bowel disease whose previous anti-TNF treatment has failed. Aliment. Pharmacol. Ther. 2015, 41, 613–623.
- Sebastian, S.; Ashton, K.; Houston, Y.; Diggory, T.M.; Dore, P. Anti-TNF therapy induced immune neutropenia in Crohns disease- report of 2 cases and review of literature. J. Crohn’s Colitis 2012, 6, 713–716.
- Biancone, L.; Armuzzi, A.; Scribano, M.L.; D’Inca, R.; Castiglione, F.; Papi, C.; Angelucci, E.; Daperno, M.; Mocciaro, F.; Riegler, G.; et al. Inflammatory Bowel Disease Phenotype as Risk Factor for Cancer in a Prospective Multicentre Nested Case-Control IG-IBD Study. J. Crohn’s Colitis 2016, 10, 913–924.
- Ford, A.C.; Peyrin-Biroulet, L. Opportunistic Infections With Anti-Tumor Necrosis Factor-α Therapy in Inflammatory Bowel Disease: Meta-Analysis of Randomized Controlled Trials. Am. J. Gastroenterol. 2013, 108, 1268–1276.
- Amiot, A.; Grimaud, J.; Peyrin-Biroulet, L.; Filippi, J.; Pariente, B.; Roblin, X.; Buisson, A.; Stefanescu, C.; Trang-Poisson, C.; Altwegg, R.; et al. Effectiveness and Safety of Vedolizumab Induction Therapy for Patients With Inflammatory Bowel Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 1593–1601.
- Schrezenmeir, J.; de Vrese, M. Probiotics, prebiotics, and synbiotics—approaching a definition. Am. J. Clin. Nutr. 2001, 73, 361s–364s.
- Gill, H.; Prasad, J. Probiotics, Immunomodulation, and Health Benefits. In Bioactive Components of Milk; Springer: New York, NY, USA, 2008; pp. 423–454.
- Kruis, W. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut 2004, 53, 1617–1623.
- Sood, A.; Midha, V.; Makharia, G.K.; Ahuja, V.; Singal, D.; Goswami, P.; Tandon, R.K. The Probiotic Preparation, VSL#3 Induces Remission in Patients With Mild-to-Moderately Active Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2009, 7, 1202–1209.
- Braat, H.; Rottiers, P.; Hommes, D.W.; Huyghebaert, N.; Remaut, E.; Remon, J.P.; van Deventer, S.J.H.; Neirynck, S.; Peppelenbosch, M.P.; Steidler, L.; et al. A Phase I Trial With Transgenic Bacteria Expressing Interleukin-10 in Crohn’s Disease. Clin. Gastroenterol. Hepatol. 2006, 4, 754–759.
- Worthley, D.L.; Le Leu, R.K.; Whitehall, V.L.; Conlon, M.; Christophersen, C.; Belobrajdic, D.; Mallitt, K.; Hu, Y.; Irahara, N.; Ogino, S.; et al. A human, double-blind, placebo-controlled, crossover trial of prebiotic, probiotic, and synbiotic supplementation: Effects on luminal, inflammatory, epigenetic, and epithelial biomarkers of colorectal cancer. Am. J. Clin. Nutr. 2009, 90, 578–586.
- Jia, K.; Tong, X.; Wang, R.; Song, X. The clinical effects of probiotics for inflammatory bowel disease. Medicine 2018, 97, e13792.
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89.
- Kelly, C.R.; Kahn, S.; Kashyap, P.; Laine, L.; Rubin, D.; Atreja, A.; Moore, T.; Wu, G. Update on Fecal Microbiota Transplantation 2015: Indications, Methodologies, Mechanisms and Outlook. Gastroenterology 2015, 149, 223–237.
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10.
- Paramsothy, S.; Paramsothy, R.; Rubin, D.T.; Kamm, M.A.; Kaakoush, N.O.; Mitchell, H.M.; Castano-Rodriguez, N. Faecal Microbiota Transplantation for Inflammatory Bowel Disease: A Systematic Review and Meta-analysis. J. Crohn’s Colitis 2017, 11, 1180–1199.
- Vaughn, B.P.; Vatanen, T.; Allegretti, J.R.; Bai, A.; Xavier, R.J.; Korzenik, J.; Gevers, D.; Ting, A.; Robson, S.C.; Moss, A.C. Increased Intestinal Microbial Diversity Following Fecal Microbiota Transplant for Active Crohnʼs Disease. Inflamm. Bowel Dis. 2016, 22, 2182–2190.
- Xiao, B.; Merlin, D. Oral colon-specific therapeutic approaches toward treatment of inflammatory bowel disease. Expert Opin. Drug Deliv. 2012, 9, 1393–1407.
- Cuvelier, C.A.; Quatacker, J.; Mielants, H.; Vos, M.d.; Veys, E.; Roels, H.J. M cells are damaged and increased in number in inflamed human ileal mucosa. Eur J. Morphol. 1993, 31, 87–91.
- Pichai, M.V. Potential prospects of nanomedicine for targeted therapeutics in inflammatory bowel diseases. World J. Gastroenterol. 2012, 18, 2895.
- Antoni, L. Intestinal barrier in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 1165.
- Han, H.-K.; Shin, H.-J.; Ha, D.H. Improved oral bioavailability of alendronate via the mucoadhesive liposomal delivery system. Eur. J. Pharm. Sci. 2012, 46, 500–507.
- Niebel, W.; Walkenbach, K.; Béduneau, A.; Pellequer, Y.; Lamprecht, A. Nanoparticle-based clodronate delivery mitigates murine experimental colitis. J. Control. Release 2012, 160, 659–665.
- Lautenschläger, C.; Schmidt, C.; Lehr, C.-M.; Fischer, D.; Stallmach, A. PEG-functionalized microparticles selectively target inflamed mucosa in inflammatory bowel disease. Eur. J. Pharm. Biopharm. 2013, 85, 578–586.
- Tirosh, B.; Khatib, N.; Barenholz, Y.; Nissan, A.; Rubinstein, A. Transferrin as a Luminal Target for Negatively Charged Liposomes in the Inflamed Colonic Mucosa. Mol. Pharm. 2009, 6, 1083–1091.
- Hua, S.; Marks, E.; Schneider, J.J.; Keely, S. Advances in oral nano-delivery systems for colon targeted drug delivery in inflammatory bowel disease: Selective targeting to diseased versus healthy tissue. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 1117–1132.
- Li, W.; Li, Y.; Liu, Z.; Kerdsakundee, N.; Zhang, M.; Zhang, F.; Liu, X.; Bauleth-Ramos, T.; Lian, W.; Makila, E.; et al. Hierarchical structured and programmed vehicles deliver drugs locally to inflamed sites of intestine. Biomaterials 2018, 185, 322–332.
- Jubeh, T.T.; Barenholz, Y.; Rubinstein, A. Differential Adhesion of Normal and Inflamed Rat Colonic Mucosa by Charged Liposomes. Pharm. Res. 2004, 21, 447–453.
- Talaei, F.; Atyabi, F.; Azhdarzadeh, M.; Dinarvand, R.; Saadatzadeh, A. Overcoming therapeutic obstacles in inflammatory bowel diseases: A comprehensive review on novel drug delivery strategies. Eur. J. Pharm. Sci. 2013, 49, 712–722.
- Wilson, D.S.; Dalmasso, G.; Wang, L.; Sitaraman, S.V.; Merlin, D.; Murthy, N. Orally delivered thioketal nanoparticles loaded with TNF-α–siRNA target inflammation and inhibit gene expression in the intestines. Nat. Mater. 2010, 9, 923–928.
- Vong, L.B.; Tomita, T.; Yoshitomi, T.; Matsui, H.; Nagasaki, Y. An Orally Administered Redox Nanoparticle That Accumulates in the Colonic Mucosa and Reduces Colitis in Mice. Gastroenterology 2012, 143, 1027–1036.
- Vong, L.B.; Yoshitomi, T.; Morikawa, K.; Saito, S.; Matsui, H.; Nagasaki, Y. Oral nanotherapeutics: Effect of redox nanoparticle on microflora in mice with dextran sodium sulfate-induced colitis. J. Gastroenterol. 2014, 49, 806–813.
- Liu, L.; Yao, W.; Rao, Y.; Lu, X.; Gao, J. pH-Responsive carriers for oral drug delivery: Challenges and opportunities of current platforms. Drug Deliv. 2017, 24, 569–581.
- Agoram, B.; Woltosz, W.S.; Bolger, M.B. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv. Drug Deliv. Rev. 2001, 50, S41–S67.
- Beloqui, A.; Coco, R.; Memvanga, P.B.; Ucakar, B.; Rieux, A.d.; Préat, V. pH-sensitive nanoparticles for colonic delivery of curcumin in inflammatory bowel disease. Int. J. Pharm. 2014, 473, 203–212.
- Barea, M.J.; Jenkins, M.J.; Gaber, M.H.; Bridson, R.H. Evaluation of liposomes coated with a pH responsive polymer. Int. J. Pharm. 2010, 402, 89–94.
- Lamprecht, A.; Yamamoto, H.; Takeuchi, H.; Kawashima, Y. Design of pH-sensitive microspheres for the colonic delivery of the immunosuppressive drug tacrolimus. Eur. J. Pharm. Biopharm. 2004, 58, 37–43.
- Lamprecht, A.; Yamamoto, H.; Ubrich, N.; Takeuchi, H.; Maincent, P.; Kawashima, Y. FK506 Microparticles Mitigate Experimental Colitis with Minor Renal Calcineurin Suppression. Pharm. Res. 2005, 22, 193–199.
- Krishnamachari, Y.; Madan, P.; Lin, S. Development of pH- and time-dependent oral microparticles to optimize budesonide delivery to ileum and colon. Int. J. Pharm. 2007, 338, 238–247.
- Makhlof, A.; Tozuka, Y.; Takeuchi, H. pH-Sensitive nanospheres for colon-specific drug delivery in experimentally induced colitis rat model. Eur. J. Pharm. Biopharm. 2009, 72, 1–8.
- Ribeiro, L.N.M.; Alcântara, A.C.S.; Darder, M.; Aranda, P.; Araújo-Moreira, F.M.; Ruiz-Hitzky, E. Pectin-coated chitosan–LDH bionanocomposite beads as potential systems for colon-targeted drug delivery. Int. J. Pharm. 2014, 463, 1–9.
- Naeem, M.; Bae, J.; Oshi, M.A.; Kim, M.; Moon, H.R.; Lee, B.L.; Im, E.; Jung, Y.; Yoo, J. Colon-targeted delivery of cyclosporine A using dual-functional Eudragit® FS30D/PLGA nanoparticles ameliorates murine experimental colitis. Int. J. Nanomed. 2018, 13, 1225–1240.
- Xu, Q.; Zhang, N.; Qin, W.; Liu, J.; Jia, Z.; Liu, H. Preparation, In Vitro and In Vivo Evaluation of Budesonide Loaded Core/Shell Nanofibers as Oral Colonic Drug Delivery System. J. Nanosci. Nanotechnol. 2013, 13, 149–156.
- Mongia, P.; Khatik, R.; Raj, R.; Jain, N.; Pathak, A.K. pH-Sensitive Eudragit S-100 Coated Chitosan Nanoparticles of 5-Amino Salicylic Acid for Colon Delivery. J. Biomater. Tissue Eng. 2014, 4, 738–743.
- Oshi, M.A.; Naeem, M.; Bae, J.; Kim, J.; Lee, J.; Hasan, N.; Kim, W.; Im, E.; Jung, Y.; Yoo, J. Colon-targeted dexamethasone microcrystals with pH-sensitive chitosan/alginate/Eudragit S multilayers for the treatment of inflammatory bowel disease. Carbohydr. Polym. 2018, 198, 434–442.
- Asghar, L.F.A.; Chandran, S. Multiparticulate Formulation Approach to Colon Specific Drug Delivery: Current Perspectives. J. Pharm Pharm Sci. 2006, 9, 327–338.
- Sharpe, L.A.; Daily, A.M.; Horava, S.D.; Peppas, N.A. Therapeutic applications of hydrogels in oral drug delivery. Expert Opin. Drug Deliv. 2014, 11, 901–915.
- Wang, Q.-S.; Wang, G.-F.; Zhou, J.; Gao, L.-N.; Cui, Y.-L. Colon targeted oral drug delivery system based on alginate-chitosan microspheres loaded with icariin in the treatment of ulcerative colitis. Int. J. Pharm. 2016, 515, 176–185.
- Oliva, N.; Conde, J.; Wang, K.; Artzi, N. Designing Hydrogels for On-Demand Therapy. Acc. Chem. Res. 2017, 50, 669–679.
- Laroui, H.; Dalmasso, G.; Nguyen, H.T.T.; Yan, Y.; Sitaraman, S.V.; Merlin, D. Drug-Loaded Nanoparticles Targeted to the Colon With Polysaccharide Hydrogel Reduce Colitis in a Mouse Model. Gastroenterology 2010, 138, 843–853.
- Chen, J.; Li, X.; Chen, L.; Xie, F. Starch film-coated microparticles for oral colon-specific drug delivery,” Carbohydr. Polym. 2018, 191, 242–254.
- Bhavsar, M.D.; Amiji, M.M. Gastrointestinal distribution and in vivo gene transfection studies with nanoparticles-in-microsphere oral system (NiMOS). J. Control. Release 2007, 119, 339–348.
- Dongowski, G.; Lorenz, A.; Anger, H. Degradation of Pectins with Different Degrees of Esterification by Bacteroides thetaiotaomicron Isolated from Human Gut Flora. Appl. Environ. Microbiol. 2000, 66, 1321–1327.
- Lee, C.-M.; Kim, D.-W.; Lee, H.-C.; Lee, K.-Y. Pectin microspheres for oral colon delivery: Preparation using spray drying method andin vitro release of indomethacin. Biotechnol. Bioprocess. Eng. 2004, 9, 191–195.
- Günter, E.A.; Markov, P.A.; Melekhin, A.K.; Belozerov, V.S.; Martinson, E.A.; Litvinets, S.G.; Popov, S.V. Preparation and release characteristics of mesalazine loaded calcium pectin-silica gel beads based on callus cultures pectins for colon-targeted drug delivery. Int. J. Biol. Macromol. 2018, 120, 2225–2233.
- Patole, V.C.; Pandit, A.P. Mesalamine-loaded alginate microspheres filled in enteric coated HPMC capsules for local treatment of ulcerative colitis: In vitro and in vivo characterization. J. Pharm. Investig. 2018, 48, 257–267.
- Iglesias, N.; Galbis, E.; Díaz-Blanco, M.; Lucas, R.; Benito, E.; De-Paz, M.-V. Nanostructured Chitosan-Based Biomaterials for Sustained and Colon-Specific Resveratrol Release. Int. J. Mol. Sci. 2019, 20, 398.
- Helmy, A.M.; Elsabahy, M.; Abd-Elkareem, M.; Ibrahim, E.A.; Soliman, G.M. High-Payload chitosan microparticles for the colonic delivery of quercetin: Development and in-vivo evaluation in a rabbit colitis model. J. Drug Deliv. Sci. Technol. 2020, 58, 101832.
- Wang, B.; Zhuang, X.; Deng, Z.; Jiang, H.; Mu, J.; Wang, Q.; Xiang, X.; Guo, H.; Zhang, L.; Dryden, G.; et al. Targeted Drug Delivery to Intestinal Macrophages by Bioactive Nanovesicles Released from Grapefruit. Mol. Ther. 2014, 22, 522–534.
- Zhang, M.; Viennois, E.; Prasad, M.; Zhang, Y.; Wang, L.; Zhang, Z.; Han, M.K.; Xiao, B.; Xu, C.; Srinivasan, S.; et al. Edible ginger-derived nanoparticles: A novel therapeutic approach for the prevention and treatment of inflammatory bowel disease and colitis-associated cancer. Biomaterials 2016, 101, 321–340.