The extracellular matrix (ECM) is the non-cellular component in the cardiac microenvironment, and serves essential structural and regulatory roles in establishing and maintaining tissue architecture and cellular function. The patterns of molecular and biochemical ECM alterations in developing and adult hearts depend on the underlying injury type and epigenetic guidelines.
- extracellular matrix
- cardiac development
- regeneration
- remodeling
- epigenetics
1. Introduction
The extracellular matrix (ECM) is made up of many proteins that hold together and direct cell adhesion and migration, as well as regulate cellular growth, metabolism and differentiation signals, and cell functions, in healthy and pathological conditions [1][2][3][4][5][6][7]. Cells that lose contact with the ECM via integrins have a higher chance of undergoing apoptosis (programmed cell death) than anchored cells. Cell adhesions intervene in effective bidirectional communications among cells and the extracellular network. ECM–cell interaction and ECM-mediated cell–cell communication play crucial roles in modulating cell adhesion, motility, survival, proliferation, differentiation, and maturation [8][9]. Utilizing integrins and non-integrin receptors (e.g., dystroglycan, sulfatides discoidin domain receptors, CD44, epidermal growth factor receptor, and P-selectin glycoprotein ligand-1) [10][11], cells can detect the physical and biochemical properties of the extracellular framework. The ECM is a highly dynamic structure present in all tissues, and maintains the structure and function of the organ, mediating the development and remodeling of the organ.
The ECM is outlined because of the cell-free elements secreted by cells that consist of macromolecules like scleroprotein, collagens, proteoglycan, hyaluronan, non-collagenous glycoproteins, and proteinases [12][13]. In the cardiac microenvironment, non-myocyte cell types populate the cardiac interstitium [14]. The heart surface is covered by epicardium, a derivative of mesothelial cells. It is termed proepicardium, for its function in giving rise to epicardium and epicardium-derived cells [15]. The cells migrate to the myocardial wall and differentiate into fibroblasts, endothelial cells, and smooth muscle cells [15]. These cells produce and release most matrix proteins, and the cell–ECM communication has an essential role in the programming and development of heart function (Figure 1). Among them, fibroblasts are the major cell type contributing to the ECM synthesis, in order to maintain the myocardial tissue architecture and mediate cell signaling through growth factor interactions and integrins [16]. Human mesenchymal stromal cells can release ECM proteins such as fibronectin (FN) and collagens into the space around cells to promote cell spreading [17]. Endothelial cells are also crucial in vascularization, cardiac function, and/or remodeling by producing ECM proteins such as collagens, laminin, elastin, fibulins, proteoglycans, matrix metalloproteinases (MMPs), tenascin-C (TNC), and thrombospodins (TSPs) [18]. In addition, immune cells in the cardiac microenvironment can also produce ECM proteins such as MMPs to modulate the immune response in the heart, contributing to the regulation of cardiomyocyte survival [19]. Therefore, cell-derived ECM and related signaling play an essential role in regulating cardiovascular function from early development to postpartum life, aging, and possibly disease.
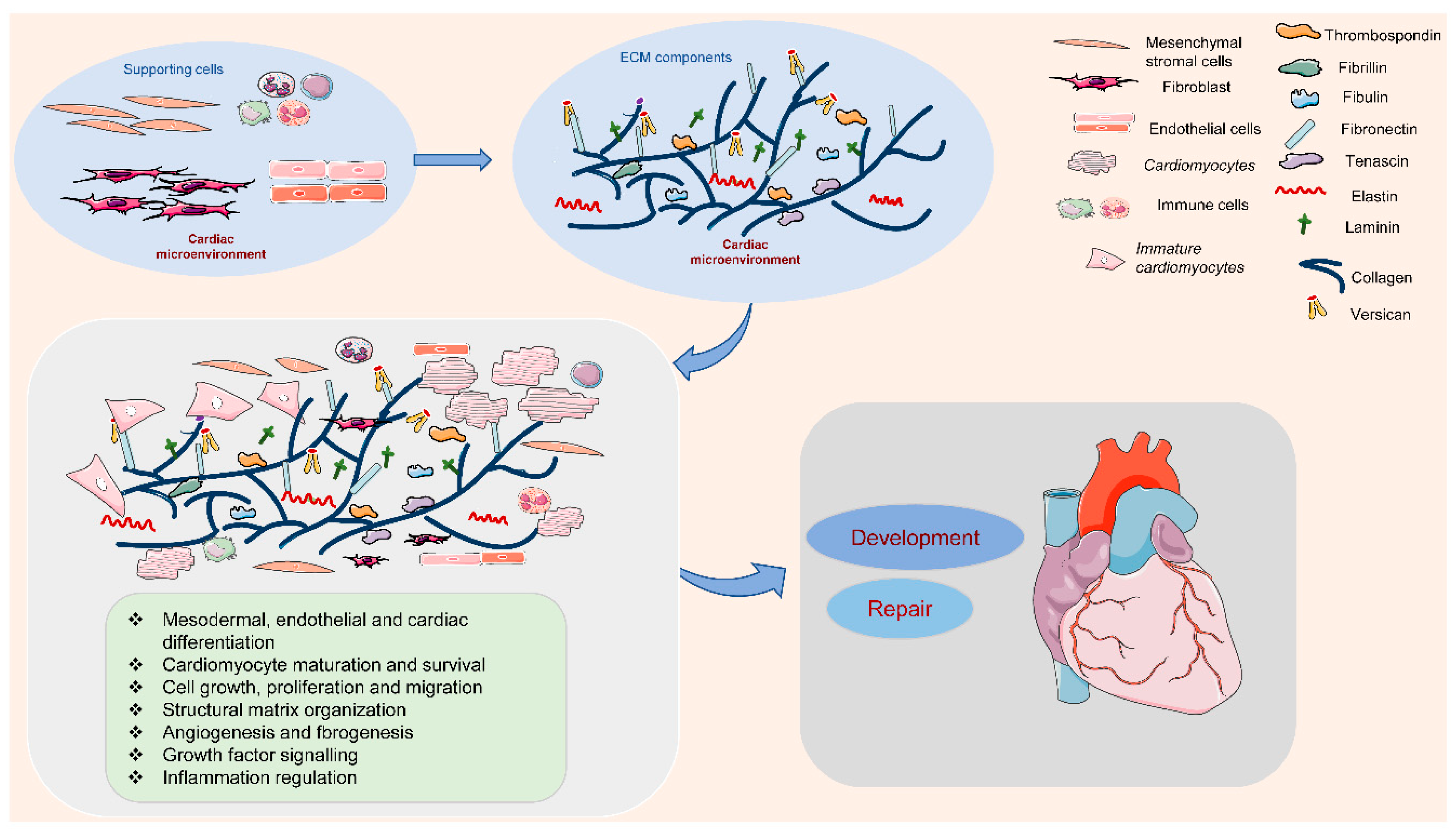
Figure 1. Extracellular matrix (ECM) components and their role in the cardiac microenvironment during heart development and repair. In the cardiac microenvironment, supporting cells, including mesenchymal stromal cells, fibroblasts, endothelial cells, and immune cells, produce the main ECM proteins. These ECM components promote cardiomyocyte differentiation, maturation, and survival, and the interaction between cardiomyocytes and supporting cells, contributing to heart development and repair.
2. The Role of ECM in Heart Development
The ECM provides essential organic components for embryogenesis and tissue maturation. The ECM is conditional; the slightest changes in its physiological state result in ruinous consequences, which might lead to severe defects or even death of the developing embryo (Table 1). In the middle development, the mechanism of dorsal closure could also be a sophisticated method, involving associate degree orchestration of cell–matrix interaction between smooth muscle cells, epithelial tissue cells, and the ECM [20]. A recent study demonstrated that nucleus–cytoskeleton–ECM connections triggered coordinated cardioblast movements, and controlled cardioblast number in Drosophila [21].
Table 1. ECM loss-of-function phenotypes in mammalian development.
|
ECM |
Isoform/ Type |
Receptor |
Phenotype |
References |
|
Fibronectin |
Integrin β1 |
Early embryonic lethality. Defects in mesodermal, neural tube, and cardiovascular development |
||
|
Laminin |
α4 |
Integrin β1, dystroglycan, and proteoglycans |
Defects in microvessel maturation, synaptic maturation |
|
|
β1 |
Integrin β1, dystroglycan, and sulfatides |
Embryonic lethality. Defects in extraembryonic tissue development, implantation, gastrulation |
[28] |
|
|
γ1 |
Integrin β1, dystroglycan, and sulfatides |
Embryonic lethality. Defects in endoderm differentiation, axonal sorting and myelination, neurite growth and neuronal migration, extraembryonic tissues development |
||
|
Collagen |
ColI |
Integrins, discoidin domain receptors 1 and 2 |
Embryonic lethality. Defects in circulatory system |
[34] |
|
ColIII |
Post-natal death. Defects in cardiovascular system and brain development |
|||
|
ColIV |
Embryonic lethality. |
|||
|
ColV |
Early embryonic lethality. Defects in fibril formation, and ventricular myocardial morphogenesis and heart valve development |
|||
|
ColXI |
Defects in skeletal morphogenesis, and ventricular myocardial morphogenesis and heart valve development |
|||
|
ColXIV |
Defects in fiber and fibril |
|||
|
ColXV |
Defects in skeletal muscle and cardiovascular development, and axonal segregation and myelination |
|||
|
Elastin |
Galectin-3, integrins, and elastin receptor complex comprising the elastin binding protein, the protective protein/cathepsin A and the membrane-bound neuramidase-1 |
Post-natal death. Defects in cardiovascular morphogenesis and development |
||
|
Fibrillin |
FBN1 |
Integrins |
Post-natal death. Defects in cardiovascular development and integrated tendon formation |
|
|
Fibulin |
Fibulin-1 |
Integrins |
Perinatal lethal. Defects in vascular, lung and kidney development |
|
|
Fibulin-4 |
Defects in elastogenesis in lungs and vasculature, and cardiovascular development |
|||
|
Fibulin-5 |
Defects in elastogenesis in the skin, lung and vasculature |
|||
|
Tenascin |
TNC |
Integrins |
Defects in neural development, alveolarization and microvascular maturation |
|
|
Versican |
CD44, integrins, epidermal growth factor receptor, and P-selectin glycoprotein ligand-1 |
Embryonic lethality. Defects in heart and neural development |
||
|
Thrombospondin |
TSP-4 |
Integrins |
Increased production of ECM and enlarged heart |
[65] |
Inherent cardiovascular disease is the leading non-infectious rationalization for death in children. It is becoming apparent that many internal organ abnormalities once thought to possess complex etiologies occur because of mutations in biological process management genes [66]. These mutations are manifested at birth as grievous internal organ malformations, or later as subtler internal organ abnormalities. Understanding the role of ECM in internal organ development has vital implications not only for an understanding inherent upset, but also for the chance of internal organ repair through genetic reprogramming of non-cardiac cells to a cardiogenic role strategic location.
The ECM gene expression profiles of embryonic and adult mouse cardiac fibroblasts revealed that higher levels of FN1, collagen genes, TNC, Postn (periostin), and Hapln1 (hyaluronan and proteoglycan link protein 1) were expressed in embryonic than adult hearts [67]. Importantly, embryonic cardiac fibroblasts promote cardiomyocyte proliferation through fibronectin and collagen, involving β1 integrin signaling, leading to myocardial growth and ventricular compaction during cardiogenesis [67]. In an environment rich in abnormal cells and growth factors, activated fibroblasts can produce matrix proteins, proteases and their inhibitors, and regulate matrix metabolism. Due to the pathological maturity, "stress shielding" of fibroblasts through the cross-linked matrix, and macromolecule withdrawal, may lead to quiescence and eventually apoptosis.
Fibulin belongs to a family of five extracellular glycoproteins and mediates the formation of proteoglycan aggregates, elastic fibers, fibronectin microfibrils, basement membrane networks, and supramolecular structures. The expression patterns of biological processes indicate that many fibrins are expressed at epithelial-mesenchymal transition sites during the entire embryogenesis, and the vascular system is related to one of these transition sites [68]. Fibulins 1 and 2 are highly expressed during cardiac valvuloseptal formation. Fibulin 1 is expressed by primordial vascular smooth muscle cells associated with the ventral endothelium of dorsal aortae and developing aortic-arch vessels [52]. In addition, fibulin 2 is expressed by coronary endothelial cells that originate from epicardial cells [69]. Interestingly, fibulin-1 deficiency, but not fibulin 2 deficiency, induced a perinatally lethal phenotype with a defective endothelial basement membrane of small vessels in mice [70]; this may be due to the functional compensation of fibulin 1.
Few studies have investigated the role of TSPs in cardiac development. Increased expression of TSP-1 in the second trimester was demonstrated to cause defects in the cardiovascular system and even embryonic lethality [71]. Conversely, the lack of TSP-4 led to increased ECM production and developmental heart enlargement [65]. As such, it is important to study the modular structures and binding interactions, and the temporal, spatial, and quantitative expression differences of various ECM proteins and their collaborations in cardiovascular health and diseases.
3. ECM in the Programming of Cardiovascular Repair
The adult heart has limited recovery and repair potential, and the loss of myocardial cells due to injury may end in heart disease and death. The cellular biological progression and restraining mechanisms associated with heart development and advancement can repair damaged adult hearts through the "stiring" pathway, which can determine the bioactivity during the entire embryogenesis. Incitement of the differentiation and proliferation of cardiomyocytes, by initiating the mitotic signaling pathway engaged with embryonic heart growth, points to a correlative methodology for heart recovery and repair [72]. Cardiac damage includes arterial sclerosis, myocardial infarction (MI), and ischemic and non-ischemic heart injury, which induces repair by the embryonic cell. Cells reply to the ECM by transforming their microenvironment, which becomes dysregulated in tube-shaped structural diseases, such as high blood pressure, restenosis, and arterial sclerosis [73].
After MI, the ECM dynamic alteration and remodeling propels inflammation and repair [74][75]. The first generation of bioactive matrix fragments activates an unhealthy signal. An extremely plastic tentative matrix formation facilitates blood corpuscle infiltration and activates infarction myofibroblasts [76]. The deposition of matrix cellular macromolecules modulates growth factor signal transduction, and promotes the spatial and temporal regulation of the repair [77]. Temporal scales vary from conformational changes in control of the particle channel gap, to fibrillation over seconds, and end in death. Spatial scales vary from metric linear unit pore sizes in membrane channels and gap junctions, to the meter length scale of the whole cardiovascular system throughout a living patient. Overwhelming changes in the ECM composition are conducive to the pathologic process of cardiac remodeling (Figure 2).
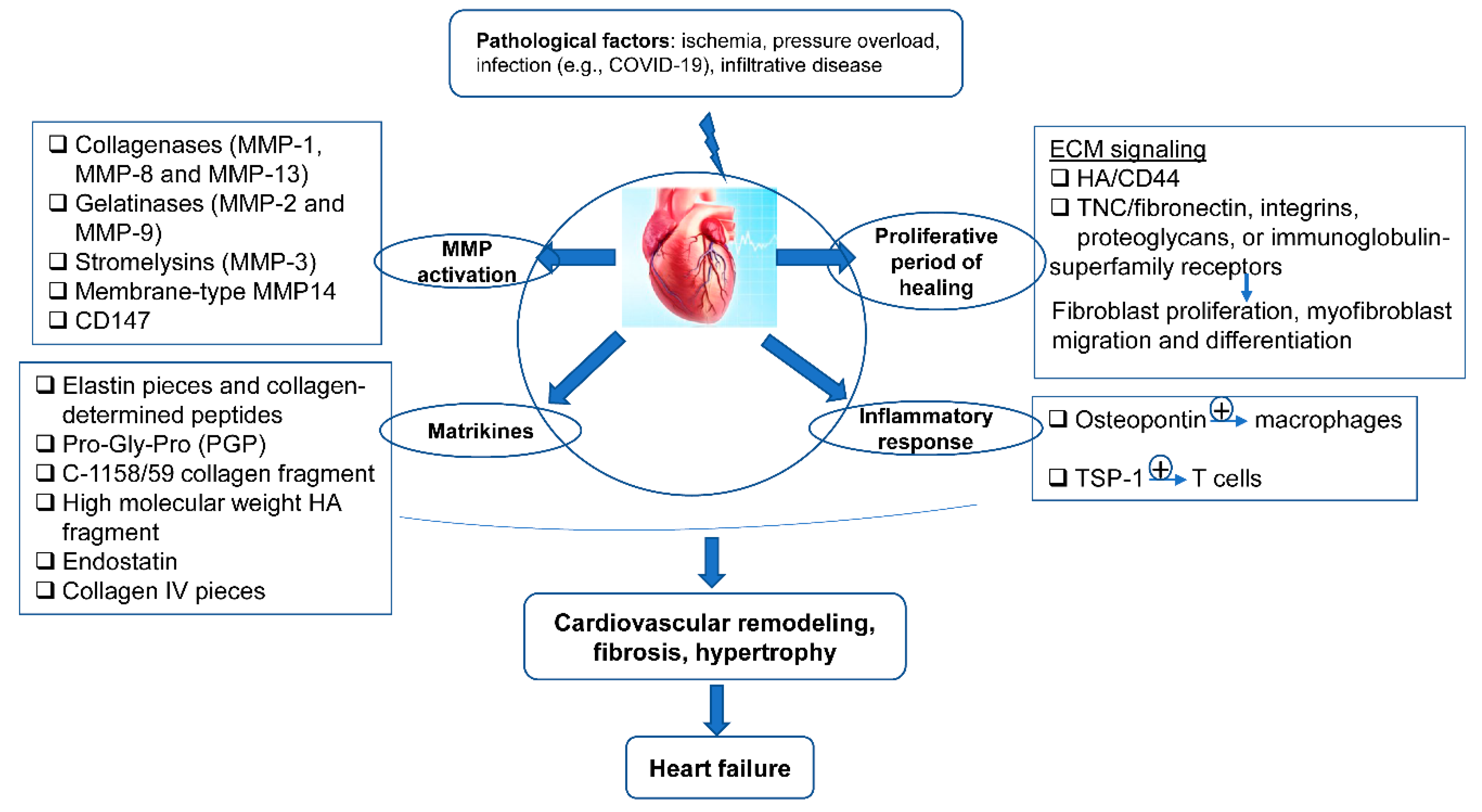
Figure 2. The ECM in the programming of cardiovascular repair and disease. Cardiovascular pathological factors induce abnormal synthesis and release of ECM proteins and ECM signaling, implicated in the process of matrix metalloproteinase (MMP) activation, matrikines production, proliferation, and inflammatory response; this results in cardiovascular remodeling, fibrosis, hypertrophy, and thus heart failure.
4. Epigenetic Regulation of ECM in Heart
Emerging research areas in the ECM field include epigenetic control of gene expression of ECM proteins, or indirectly, by modulating the expression of genes that regulate the synthesis or the degradation of ECM molecules in development and disease onset. Epigenetic changes characterized by RNA and DNA methylation, non-coding RNAs-intervened quality guidelines, and histone adjustments, have been seen in cardiovascular dysfunction and heart recovery (Figure 3), yet the components are indistinct. Knowledge of these aspects will deepen our understanding of ECM regulatory roles in cardiac health and disease, and inform new pharmacological agents targeting ECM-related cardiovascular diseases.
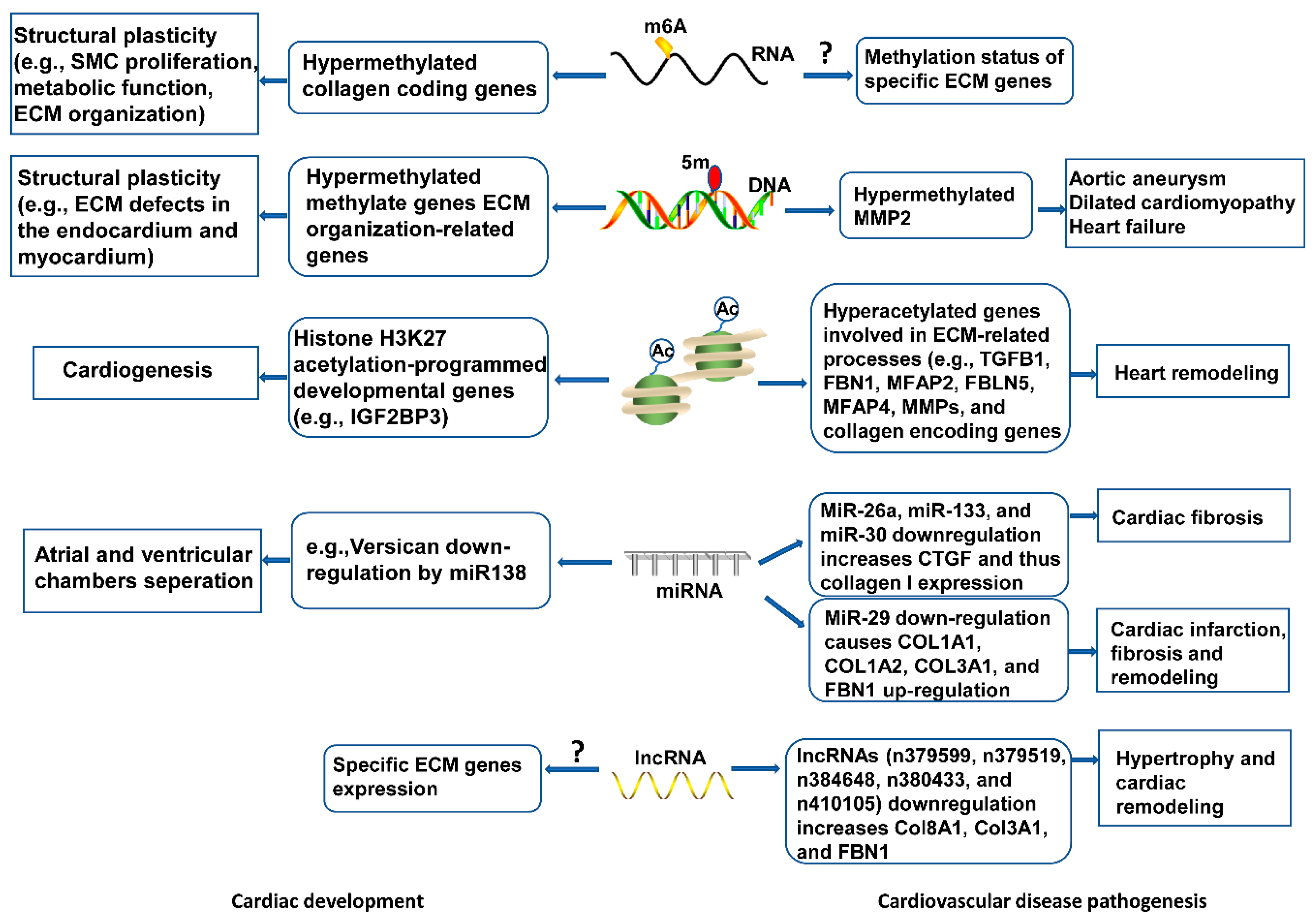
Figure 3. Epigenetic mechanisms of ECM modulation in cardiovascular development and disease. Epigenetic events, including RNA and DNA methylation, histone modifications, and non-coding RNAs mediate ECM gene expression, contributing to cytoskeletal architecture, remodeling, and functional response in heart development. Abnormal epigenetic modification may disrupt ECM homeostasis, leading to cardiovascular pathogenesis.
5. Conclusions
The ECM is a crucial element of the heart. Regulation of ECM structural integrity influences the viscus structure and performance. The strict regulation of temporal and spatial expression, and the proteolytic processing of ECM elements by extracellular proteases are crucial for the development of traditional internal organs. ECM pathological transformation is commonly related to viscus pathology alternative adverse outcomes, while the physiological turnover of ECM is beneficial for the process of tissue regeneration and repair.
Imperfect development in the womb is related to the tendency for cardiovascular disease in adulthood, an idea named "developmental origins of health and disease". More and more evidence supports the association of epigenetic guidelines with the underlying mechanism. Epigenetic systems, for example, RNA and DNA methylation, histone adjustments, and non-coding RNAs, give a degree of quality guidelines without modifying DNA arrangements. These changes are moderately steady signals, offering possible knowledge into the instrument's fundamental formative starting points of wellbeing and ailment. Therefore, it is imperative to understand the underlying mechanisms of ECM regulation that control cardiovascular development. Understanding the developmental mechanisms of ECM regulation could contribute to developing therapeutic strategies for cardiovascular disease.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21228610
References
- Loganathan, R.; Rongish, B.J.; Smith, C.M.; Filla, M.B.; Czirok, A.; Bénazéraf, B.; Little, C.D. Extracellular matrix motion and early morphogenesis. Development 2016, 143, 2056–2065, doi:10.1242/dev.127886.
- Smith, L.R.; Cho, S.; Discher, D.E. Stem Cell Differentiation is Regulated by Extracellular Matrix Mechanics. Physiol. 2018, 33, 16–25, doi:10.1152/physiol.00026.2017.
- Arseni, L.; Lombardi, A.; Orioli, D. From Structure to Phenotype: Impact of Collagen Alterations on Human Health. Int. J. Mol. Sci. 2018, 19, 1407, doi:10.3390/ijms19051407.
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99, doi:10.1016/j.mam.2018.07.001.
- Rickard, A.J.; Morgan, J.; Bienvenu, L.A.; Fletcher, E.K.; Cranston, G.A.; Shen, J.Z.; Reichelt, M.E.; Delbridge, L.M.; Young, M.J. Cardiomyocyte Mineralocorticoid Receptors Are Essential for Deoxycorticosterone/Salt-Mediated Inflammation and Cardiac Fibrosis. Hypertens. 2012, 60, 1443–1450, doi:10.1161/hypertensionaha.112.203158.
- McCain, M.L.; Agarwal, A.; Nesmith, H.W.; Nesmith, A.P.; Parker, K.K. Micromolded gelatin hydrogels for extended culture of engineered cardiac tissues. Biomater. 2014, 35, 5462–5471, doi:10.1016/j.biomaterials.2014.03.052.
- Russo, I.; Cavalera, M.; Huang, S.; Su, Y.; Hanna, A.; Chen, B.; Shinde, A.V.; Conway, S.J.; Graff, J.M.; Frangogiannis, N.G. Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program. Circ. Res. 2019, 124, 1214–1227, doi:10.1161/circresaha.118.314438.
- Ross, R.S.; Borg, T.K. Integrins and the Myocardium. Circ. Res. 2001, 88, 1112–1119, doi:10.1161/hh1101.091862.
- Sa, S.; Wong, L.; McCloskey, K.E. Combinatorial Fibronectin and Laminin Signaling Promote Highly Efficient Cardiac Differentiation of Human Embryonic Stem Cells. BioResearch Open Access 2014, 3, 150–161, doi:10.1089/biores.2014.0018.
- Gullberg, D.; Ekblom, P. Extracellular matrix and its receptors during development. Int. J. Dev. Biol. 1995, 39, 845–854.
- Cichy, J.; PuréE. The liberation of CD44. J. Cell Biol. 2003, 161, 839–843, doi:10.1083/jcb.200302098.
- Jarvelainen, H.; Sainio, A.; Koulu, M.; Wight, T.N.; Penttinen, R., Extracellular matrix molecules: potential targets in pharmacotherapy. Pharm. Rev. 2009, 61, 198–223.
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123 (Pt 24), 4195–4200, doi:10.1242/jcs.023820.
- Varga, I.; Kyselovič, J.; Gálfiová, P.; Danisovic, L. The Non-cardiomyocyte Cells of the Heart. Their Possible Roles in Exercise-Induced Cardiac Regeneration and Remodeling. Advances in Experimental Medicine and Biology 2017, 999, 117–136, doi:10.1007/978-981-10-4307-9_8.
- Günthel, M.; Barnett, P.; Christoffels, V.M. Development, Proliferation, and Growth of the Mammalian Heart. Mol. Ther. 2018, 26, 1599–1609, doi:10.1016/j.ymthe.2018.05.022.
- Hortells, L.; Johansen, A.K.Z.; Yutzey, K.E. Cardiac Fibroblasts and the Extracellular Matrix in Regenerative and Nonregenerative Hearts. J. Cardiovasc. Dev. Dis. 2019, 6, 29, doi:10.3390/jcdd6030029.
- Horton, E.R.; Vallmajo‐Martin, Q.; Martin, I.; Snedeker, J.G.; Ehrbar, M.; Blache, U. Extracellular Matrix Production by Mesenchymal Stromal Cells in Hydrogels Facilitates Cell Spreading and Is Inhibited by FGF‐2. Adv. Heal. Mater. 2020, 9, e1901669, doi:10.1002/adhm.201901669.
- Segers, V.F.M.; Brutsaert, D.L.; De Keulenaer, G.W. Cardiac Remodeling: Endothelial Cells Have More to Say Than Just NO. Front. Physiol. 2018, 9, 382, doi:10.3389/fphys.2018.00382.
- Alfonso-Jaume, M.A.; Bergman, M.R.; Mahimkar, R.; Cheng, S.; Jin, Z.Q.; Karliner, J.S.; Lovett, D.H. Cardiac ischemia-reperfusion injury induces matrix metalloproteinase-2 expression through the AP-1 components FosB and JunB. Am. J. Physiol. Circ. Physiol. 2006, 291, H1838–H1846, doi:10.1152/ajpheart.00026.2006.
- Goodwin, K.; Lostchuck, E.E.; Cramb, K.M.L.; Zulueta-Coarasa, T.; Fernandez-Gonzalez, R.; Tanentzapf, G. Cell–cell and cell–extracellular matrix adhesions cooperate to organize actomyosin networks and maintain force transmission during dorsal closure. Mol. Biol. Cell 2017, 28, 1301–1310, doi:10.1091/mbc.e17-01-0033.
- Dondi, C.; Bertin, B.; Daponte, J.-P.; Wojtowicz, I.; Jagla, K.; Junion, G. A polarized nucleus-cytoskeleton-ECM connection controls collective migration and cardioblasts number in Drosophila. BioRxiv 2019, 1–18, doi:10.1101/2019.12.17.879932.
- George, E.L.; Georges-Labouesse, E.N.; Patel-King, R.S.; Rayburn, H.; Hynes, R. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development 1993, 119, 1079–1091.
- Cheng, P.; Andersen, P.; Hassel, D.; Kaynak, B.L.; Limphong, P.; Juergensen, L.; Kwon, C.; Srivastava, D., Fibronectin mediates mesendodermal cell fate decisions. Development 2013, 140, 2587–2596.
- De Almeida, P.G.; Pinheiro, G.G.; Nunes, A.M.; Gonçalves, A.B.; Thorsteinsdóttir, S. Fibronectin assembly during early embryo development: A versatile communication system between cells and tissues. Dev. Dyn. 2016, 245, 520–535, doi:10.1002/dvdy.24391.
- Thyboll, J.; Kortesmaa, J.; Cao, R.; Soininen, R.; Wang, L.; Iivanainen, A.; Sorokin, L.; Risling, M.; Cao, Y.; Tryggvason, K. Deletion of the Laminin α4 Chain Leads to Impaired Microvessel Maturation. Mol. Cell. Biol. 2002, 22, 1194–1202, doi:10.1128/mcb.22.4.1194-1202.2002.
- Patton, B.L.; Cunningham, J.M.; Thyboll, J.; Kortesmaa, J.; Westerblad, H.; Edström, L.; Tryggvason, K.; Sanes, J.R. Properly formed but improperly localized synaptic specializations in the absence of laminin α4. Nat. Neurosci. 2001, 4, 597–604, doi:10.1038/88414.
- Wang, J.; Hoshijima, M.; Lam, J.; Zhou, Z.; Jokiel, A.; Dalton, N.D.; Hultenby, K.; Ruiz-Lozano, P.; Ross, J.; Tryggvason, K.; et al. Cardiomyopathy Associated with Microcirculation Dysfunction in Laminin α4 Chain-deficient Mice. J. Biol. Chem. 2006, 281, 213–220, doi:10.1074/jbc.m505061200.
- Miner, J.H.; Li, C.; Mudd, J.L.; Go, G.; Sutherland, A.E. Compositional and structural requirements for laminin and basement membranes during mouse embryo implantation and gastrulation. Development 2004, 131, 2247–2256, doi:10.1242/dev.01112.
- Smyth, N.; Vatansever, H.S.; Murray, P.; Meyer, M.; Frie, C.; Paulsson, M.; Edgar, D. Absence of Basement Membranes after Targeting the LAMC1 Gene Results in Embryonic Lethality Due to Failure of Endoderm Differentiation. J. Cell Biol. 1999, 144, 151–160, doi:10.1083/jcb.144.1.151.
- Chen, Z.L.; Strickland, S., Laminin gamma1 is critical for Schwann cell differentiation, axon myelination, and regeneration in the peripheral nerve. J. Cell Biol. 2003, 163, 889–899.
- Chen, Z.L.; Haegeli, V.; Yu, H.; Strickland, S., Cortical deficiency of laminin gamma1 impairs the AKT/GSK-3beta signaling pathway and leads to defects in neurite outgrowth and neuronal migration. Dev. Biol. 2009, 327, 158–168.
- Kiyozumi, D.; Taniguchi, Y.; Nakano, I.; Toga, J.; Yagi, E.; Hasuwa, H.; Ikawa, M.; Sekiguchi, K., Laminin gamma1 C-terminal Glu to Gln mutation induces early postimplantation lethality. Life Sci. Alliance 2018, 1, e201800064.
- McKee, K.K.; Yang, D.H.; Patel, R.; Chen, Z.L.; Strickland, S.; Takagi, J.; Sekiguchi, K.; Yurchenco, P.D., Schwann cell myelination requires integration of laminin activities. J. Cell Sci. 2012, 125, 4609–4619.
- Löhler, J.; Timpl, R.; Jaenisch, R. Embryonic lethal mutation in mouse collagen I gene causes rupture of blood vessels and is associated with
- Liu, X.; Wu, H.; Byrne, M.; Krane, S.; Jaenisch, R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 1852–1856.
- Vandervore, L.; Stouffs, K.; Tanyalçin, I.; Vanderhasselt, T.; Roelens, F.; Holder-Espinasse, M.; Jørgensen, A.; Pepin, M.G.; Petit, F.; Van Kien, P.K.; et al. Bi-allelic variants inCOL3A1encoding the ligand to GPR56 are associated with cobblestone-like cortical malformation, white matter changes and cerebellar cysts. J. Med. Genet. 2017, 54, 432–440, doi:10.1136/jmedgenet-2016-104421.
- Poschl, E.; Schlötzer-Schrehardt, U.; Brachvogel, B.; Saito, K.; Ninomiya, Y.; Mayer, U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Dev. 2004, 131, 1619–1628, doi:10.1242/dev.01037.
- Abrahamson, D.R.; Hudson, B.G.; Stroganova, L.; Borza, D.-B.; John, P.L.S. Cellular Origins of Type IV Collagen Networks in Developing Glomeruli. J. Am. Soc. Nephrol. 2009, 20, 1471–1479, doi:10.1681/asn.2008101086.
- Wenstrup, R.J.; Florer, J.B.; Brunskill, E.W.; Bell, S.M.; Chervoneva, I.; Birk, D.E. Type V Collagen Controls the Initiation of Collagen Fibril Assembly. J. Biol. Chem. 2004, 279, 53331–53337, doi:10.1074/jbc.m409622200.
- Sun, M.; Chen, S.; Adams, S.M.; Florer, J.B.; Liu, H.; Kao, W.W.-Y.; Wenstrup, R.J.; Birk, D.E. Collagen V is a dominant regulator of collagen fibrillogenesis: dysfunctional regulation of structure and function in a corneal-stroma-specific Col5a1-null mouse model. J. Cell Sci. 2011, 124, 4096–4105, doi:10.1242/jcs.091363.
- Lincoln, J.; Florer, J.B.; Deutsch, G.H.; Wenstrup, R.J.; Yutzey, K.E. ColVa1 and ColXIa1 are required for myocardial morphogenesis and heart valve development. Dev. Dyn. 2006, 235, 3295–3305, doi:10.1002/dvdy.20980.
- Li, Y.; Lacerda, D.; Warman, M.; Beier, D.; Yoshioka, H.; Ninomiya, Y.; Oxford, J.; Morris, N.; Andrikopoulos, K.; Ramirez, F.; et al. A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell 1995, 80, 423–430, doi:10.1016/0092-8674(95)90492-1.
- Ansorge, H.L.; Meng, X.; Zhang, G.; Veit, G.; Sun, M.; Klement, J.F.; Beason, D.P.; Soslowsky, L.J.; Koch, M.; Birk, D.E. Type XIV Collagen Regulates Fibrillogenesis. J. Biol. Chem. 2009, 284, 8427–8438, doi:10.1074/jbc.m805582200.
- Tao, G.; Levay, A.K.; Peacock, J.D.; Huk, D.J.; Both, S.N.; Purcell, N.H.; Pinto, J.R.; Galantowicz, M.L.; Koch, M.; Lucchesi, P.A.; et al. Collagen XIV is important for growth and structural integrity of the myocardium. J. Mol. Cell. Cardiol. 2012, 53, 626–638, doi:10.1016/j.yjmcc.2012.08.002.
- Eklund, L.; Piuhola, J.; Komulainen, J.; Sormunen, R.; Ongvarrasopone, C.; Fassler, R.; Muona, A.; Ilves, M.; Ruskoaho, H.; Takala, T.E.; et al. Lack of type XV collagen causes a skeletal myopathy and cardiovascular defects in mice. Proc. Natl Acad Sci U S A 2001, 98, 1194–1199.
- Rasi, K.; Hurskainen, M.; Kallio, M.; Stavén, S.; Sormunen, R.; Heape, A.M.; Avila, R.L.; Kirschner, D.A.; Muona, A.; Tolonen, U.; et al. Lack of Collagen XV Impairs Peripheral Nerve Maturation and, When Combined with Laminin-411 Deficiency, Leads to Basement Membrane Abnormalities and Sensorimotor Dysfunction. J. Neurosci. 2010, 30, 14490–14501, doi:10.1523/jneurosci.2644-10.2010.
- Li, D.Y.; Brooke, B.; Davis, E.C.; Mecham, R.P.; Sorensen, L.K.; Boak, B.B.; Eichwald, E.; Keating, M.T. Elastin is an essential determinant of arterial morphogenesis. Nat. Cell Biol. 1998, 393, 276–280, doi:10.1038/30522.
- Wagenseil, J.E.; Ciliberto, C.H.; Knutsen, R.H.; Levy, M.A.; Kovacs, A.; Mecham, R.P. Reduced Vessel Elasticity Alters Cardiovascular Structure and Function in Newborn Mice. Circ. Res. 2009, 104, 1217–1224, doi:10.1161/CIRCRESAHA.108.192054.
- Lin, C.-J.; Staiculescu, M.C.; Hawes, J.Z.; Cocciolone, A.J.; Hunkins, B.M.; Roth, R.A.; Lin, C.-Y.; Mecham, R.P.; Wagenseil, J.E. Heterogeneous Cellular Contributions to Elastic Laminae Formation in Arterial Wall Development. Circ. Res. 2019, 125, 1006–1018, doi:10.1161/circresaha.119.315348.
- Pereira, L.; Andrikopoulos, K.; Tian, J.; Lee, S.Y.; Keene, D.R.; Ono, R.N.; Reinhardt, D.P.; Sakai, L.Y.; Biery, N.J.; Bunton, T.; et al. Targetting of the gene encoding fibrillin–1 recapitulates the vascular aspect of Marfan syndrome. Nat. Genet. 1997, 17, 218–222, doi:10.1038/ng1097-218.
- Tran, P.H.T.; Skrba, T.; Wondimu, E.; Galatioto, G.; Svensson, R.B.; Olesen, A.T.; Mackey, A.L.; Magnusson, S.P.; Ramirez, F.; Kjaer, M. The influence of fibrillin-1 and physical activity upon tendon tissue morphology and mechanical properties in mice. Physiol. Rep. 2019, 7, e14267, doi:10.14814/phy2.14267.
- Kostka, G.; Giltay, R.; Bloch, W.; Addicks, K.; Timpl, R.; FässlerR.; Chu, M.-L. Perinatal Lethality and Endothelial Cell Abnormalities in Several Vessel Compartments of Fibulin-1-Deficient Mice. Mol. Cell. Biol. 2001, 21, 7025–7034, doi:10.1128/mcb.21.20.7025-7034.2001.
- Ito, S.; Yokoyama, U.; Nakakoji, T.; Cooley, M.A.; Sasaki, T.; Hatano, S.; Kato, Y.; Saito, J.; Nicho, N.; Iwasaki, S.; et al. Fibulin-1 Integrates Subendothelial Extracellular Matrices and Contributes to Anatomical Closure of the Ductus Arteriosus. Arter. Thromb. Vasc. Biol. 2020, 40, 2212–2226, doi:10.1161/atvbaha.120.314729.
- McLaughlin, P.J.; Chen, Q.; Horiguchi, M.; Starcher, B.C.; Stanton, J.B.; Broekelmann, T.J.; Marmorstein, A.D.; McKay, B.; Mecham, R.; Nakamura, T.; et al. Targeted Disruption of Fibulin-4 Abolishes Elastogenesis and Causes Perinatal Lethality in Mice. Mol. Cell. Biol. 2006, 26, 1700–1709, doi:10.1128/mcb.26.5.1700-1709.2006.
- Igoucheva, O.; Alexeev, V.; Halabi, C.M.; Adams, S.M.; Stoilov, I.; Sasaki, T.; Arita, M.; Donahue, A.; Mecham, R.P.; Birk, D.E.; et al. Fibulin-4 E57K Knock-in Mice Recapitulate Cutaneous, Vascular and Skeletal Defects of Recessive Cutis Laxa 1B with both Elastic Fiber and Collagen Fibril Abnormalities. J. Biol. Chem. 2015, 290, 21443–21459, doi:10.1074/jbc.m115.640425.
- Halabi, C.M.; Broekelmann, T.J.; Lin, M.; Lee, V.; Chu, M.-L.; Mecham, R.P. Fibulin-4 is essential for maintaining arterial wall integrity in conduit but not muscular arteries. Sci. Adv. 2017, 3, e1602532, doi:10.1126/sciadv.1602532.
- 57. Nakamura, T.; Lozano, P.R.; Ikeda, Y.; Iwanaga, Y.; Hinek, A.; Minamisawa, S.; Cheng, C.F.; Kobuke, K.; Dalton, N.; Takada, Y.; et al. Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature 2002, 415, (6868), 171–175.
- Choi, J.; Bergdahl, A.; Zheng, Q.; Starcher, B.; Yanagisawa, H.; Davis, E.C. Analysis of dermal elastic fibers in the absence of fibulin-5 reveals potential roles for fibulin-5 in elastic fiber assembly. Matrix Biol. 2009, 28, 211–220, doi:10.1016/j.matbio.2009.03.004.
- Fukamauchi, F.; Mataga, N.; Wang, Y.-J.; Sato, S.; Yoshiki, A.; Kusakabe, M. Abnormal Behavior and Neurotransmissions of Tenascin Gene Knockout Mouse. Biochem. Biophys. Res. Commun. 1996, 221, 151–156, doi:10.1006/bbrc.1996.0561.
- Mund, S.I.; Schittny, J.C. Tenascin-C deficiency impairs alveolarization and microvascular maturation during postnatal lung development. J. Appl. Physiol. 2020, 128, 1287–1298, doi:10.1152/japplphysiol.00258.2019.
- Gottschling, C.; Wegrzyn, D.; Denecke, B.; Faissner, A. Elimination of the four extracellular matrix molecules tenascin-C, tenascin-R, brevican and neurocan alters the ratio of excitatory and inhibitory synapses. Sci. Rep. 2019, 9, 1–17, doi:10.1038/s41598-019-50404-9.
- Mjaatvedt, C.; Yamamura, H.; Capehart, A.; Turner, D.; Markwald, R. TheCspg2Gene, Disrupted in thehdfMutant, Is Required for Right Cardiac Chamber and Endocardial Cushion Formation. Dev. Biol. 1998, 202, 56–66, doi:10.1006/dbio.1998.9001.
- Kern, C.B.; Norris, R.A.; Thompson, R.P.; Argraves, W.S.; Fairey, S.E.; Reyes, L.; Hoffman, S.; Markwald, R.R.; Mjaatvedt, C.H. Versican proteolysis mediates myocardial regression during outflow tract development. Dev. Dyn. 2007, 236, 671–683, doi:10.1002/dvdy.21059.
- Stigson, M.; Löfberg, J.; Kjellén, L. Reduced Epidermal Expression of a PG-M/Versican-like Proteoglycan in Embryos of the White Mutant Axolotl. Exp. Cell Res. 1997, 236, 57–65, doi:10.1006/excr.1997.3702.
- Frolova, E.G.; Drazba, J.; Krukovets, I.; Kostenko, V.; Blech, L.; Harry, C.; Vasanji, A.; Drumm, C.; Sul, P.; Jenniskens, G.J.; et al. Control of organization and function of muscle and tendon by thrombospondin-4. Matrix Biol. 2014, 37, 35–48, doi:10.1016/j.matbio.2014.02.003.
- Jackson, M.; Marks, L.; May, G.H.; Wilson, J.B. The genetic basis of disease. Essays Biochem. 2018, 62, 643–723, doi:10.1042/ebc20170053.
- Ieda, M.; Tsuchihashi, T.; Ivey, K.N.; Ross, R.S.; Hong, T.T.; Shaw, R.M.; Srivastava, D., Cardiac fibroblasts regulate myocardial proliferation through beta1 integrin signaling. Dev. Cell 2009, 16, 233–244.
- Chu, M.-L.; Tsuda, T. Fibulins in development and heritable disease. Birth Defects Res. Part. C Embryo Today: Rev. 2004, 72, 25–36, doi:10.1002/bdrc.20003.
- Tsuda, T.; Wang, H.; Timpl, R.; Chu, M.-L. Fibulin-2 expression marks transformed mesenchymal cells in developing cardiac valves, aortic arch vessels, and coronary vessels. Dev. Dyn. 2001, 222, 89–100, doi:10.1002/dvdy.1172.
- Sicot, F.-X.; Tsuda, T.; Markova, D.; Klement, J.F.; Arita, M.; Zhang, R.-Z.; Pan, T.-C.; Mecham, R.P.; Birk, D.E.; Chu, M.-L. Fibulin-2 Is Dispensable for Mouse Development and Elastic Fiber Formation. Mol. Cell. Biol. 2007, 28, 1061–1067, doi:10.1128/mcb.01876-07.
- Fouladkou, F.; Lu, C.; Jiang, C.; Zhou, L.; She, Y.; Walls, J.R.; Kawabe, H.; Brose, N.; Henkelman, R.M.; Huang, A.; et al. The Ubiquitin Ligase Nedd4-1 Is Required for Heart Development and Is a Suppressor of Thrombospondin-1. J. Biol. Chem. 2009, 285, 6770–6780, doi:10.1074/jbc.m109.082347.
- Xin, M.; Olson, E.N.; Bassel-Duby, R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. Cell Biol. 2013, 14, 529–541, doi:10.1038/nrm3619.
- Matsumura, G.; Isayama, N.; Matsuda, S.; Taki, K.; Sakamoto, Y.; Ikada, Y.; Yamazaki, K. Long-term results of cell-free biodegradable scaffolds for in situ tissue engineering of pulmonary artery in a canine model. Biomater. 2013, 34, 6422–6428, doi:10.1016/j.biomaterials.2013.05.037.
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612, doi:10.1172/jci87491.
- Frangogiannis, N.G. The Extracellular Matrix in Ischemic and Nonischemic Heart Failure. Circ. Res. 2019, 125, 117–146, doi:10.1161/circresaha.119.311148.
- Terajima, Y.; Shimizu, T.; Tsuruyama, S.; Sekine, H.; Ishii, H.; Yamazaki, K.; Hagiwara, N.; Okano, T. Autologous Skeletal Myoblast Sheet Therapy for Porcine Myocardial Infarction Without Increasing Risk of Arrhythmia. Cell Med. 2013, 6, 99–109, doi:10.3727/215517913X672254.
- Frangogiannis, N.G.; Ren, G.; Dewald, O.; Zymek, P.; Haudek, S.; Koerting, A.; Winkelmann, K.; Michael, L.H.; Lawler, J.; Entman, M.L. Critical Role of Endogenous Thrombospondin-1 in Preventing Expansion of Healing Myocardial Infarcts. Circulation 2005, 111, 2935–2942, doi:10.1161/circulationaha.104.510354.