Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Activating transcription factor 4 (ATF4) is a transcription factor known to regulate genes associated with the sensing of cellular stress such as amino acid deprival, protein misfolding, growth arrest, and cell death. Despite its key role at the crossroads of immune and stress responses, the precise impact of ATF4 during viral infections remains unclear. Thus, ATF4 has a dual role in promoting cell survival or cell death, but also in limiting infection or participating in viral replication.
- ISR
- AIDS
- immunity
- mitochondria
- ER stress
- UPR
1. Introduction
One of the main regulators of cellular homeostasis is activating transcription factor 4 (ATF4). This bZIP domain transcription factor plays a crucial role in both the integrated stress response (ISR) and the mitochondrial stress response (MSR) [1,2,3]. In reaction to oxidative stress, misfolded protein accumulation, or nutrient deprivation, ATF4 triggers the expression of genes involved in cellular processes participating in the control of autophagy or cell death. Furthermore, the role of ATF4 in cellular responses to stress is intimately linked to its dimerization partners such as the transcription factor ATF5, an ATF4 paralogue found in mammals, which belongs to the ATF4 bZIP-domain transcription factor family [4,5]. ATF5 is also strongly involved in stress responses and can be a target of ATF4. Thus, ATF4 and ATF5 are key cellular factors that integrate various stress signals.
2. HIV-1 Infection Regulates ATF4
2.1. ATF4 Is Up-Regulated during HIV-1 and SIV Infections
HIV-1 infection leads to the development of acquired immunodeficiency syndrome (AIDS) associated with the depletion of CD4+ T cells, mainly by apoptosis [65,66,67] which predicts further pathogenicity [68,69]. Of interest, the induction of ATF4 following HIV-1 infection has been observed in several in vitro models. It was first described in Jurkat T cells, in which ATF4 was up-regulated at both the transcript and protein levels 8 h post-infection and remained elevated 48 h later [6]. This increase in ATF4 transcript levels was further observed in primary human CD4+ T cells at day 5 post-infection [8]. In a model of HIV-1 latency, ATF4 transcript and protein levels are weakly detectable [10] but increased with viral reactivation [6,8,10]. In vivo, in monkeys infected with simian immunodeficiency virus (SIV), ATF4 transcripts are also up-regulated in the gut mucosa. This increase occurred during the acute phase of SIV infection, i.e., 1 to 2 weeks post-infection, but are not observed during the chronic phase [8]. Viral proteins released in the microenvironment of infected cells such as Tat has been proposed to be sufficient for inducing ATF4 gene expression through ER stress in non-infected cells [11]. Altogether, these observations show that ATF4 expression is differentially regulated during both the early and chronic phases of viral replication, in infected but also non-infected cells, which are close to the former, suggesting that ATF4 induction is related to viral stress and plays a potential role in the establishment of latency.
2.2. How Can HIV-1 Regulate ATF4?
2.2.1. HIV-1-Induced ISR/ATF4 Signaling
The ISR leads to a global translation blockade through the phosphorylation of eIF2α, which increases ATF4 translation [3]. Several kinases are responsible for eIF2α phosphorylation: double-stranded RNA-dependent protein kinase (PKR), PKR-like ER kinase (PERK), general control non-derepressible 2 (GCN2), heme-regulated eIF2α kinase (HRI), microtubule affinity-regulating kinase 2 (MARK2), and family with sequence similarity 69 member C (FAM69C) [3,70,71] (Figure 1). The role of eIF2α and its kinases during viral infection has been recently reviewed by Liu et al. [72]. We focus on HIV-1 studies providing evidence that eIF2α phosphorylation by ISR kinases can be associated with ATF4 induction, and that a direct control of these kinases by HIV-1 proteins may affect ATF4 activity.
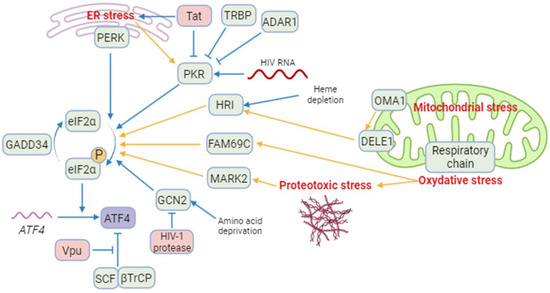
Figure 1. A model of regulation of ATF4 and integrated stress kinases during HIV-1 infection. Blue arrows correspond to interactions demonstrated by the literature in the context of HIV-1 infection. Yellow arrows correspond to reports in a different context. Besides ATF4, cellular proteins are indicated in green and HIV-1 proteins in red. ISR kinases can be activated by ER stress (PERK and PKR), HIV-1 RNA (PKR), heme depletion and cleavage of DELE1 by OMA1 after mitochondrial stress (HRI), amino acid deprival (GCN2), oxidative stress (FAM69C), and proteotoxic stress (MARK2). Several cellular and viral proteins modulate ISR kinases during HIV-1 infection, as TRBP and ADAR1 and the HIV-1 Tat protein preventing the phosphorylation of eIF2α and increasing ATF4 synthesis. The viral Vpu protein stabilizes ATF4 by opposing its ubiquitination by the SCF-βTrCP complex. Created with BioRender.com (accessed on 18 January 2024).
Among the ISR kinases, PKR is the best characterized viral nucleic acid sensor [73,74]. It is activated mainly by double-stranded RNAs (dsRNA) [75,76,77,78]. PKR regulates HIV-1 infection as it can bind the hairpin structure within the transactivation-response region of the HIV-1 genome [79]. While PKR contributes to ATF4 translation in response to endoplasmic reticulum (ER) stress [80], paradoxically, to our knowledge, no study reports the induction of ATF4 in the context of HIV-1 infection through an endogenous PKR/eIF2α pathway. A reason that may explain this lack of evidence is the rapid inhibition of PKR by multiple mechanisms during HIV-1 replication [79] (Figure 1): PKR activation is inhibited by high levels of dsRNA and by the direct binding of cellular proteins, including TAR RNA-binding protein (TRBP) and adenosine deaminase acting on RNA (ADAR1) [81,82,83,84,85,86,87]. Moreover, the viral Tat protein may counteract PKR activation by preventing PKR autophosphorylation. Tat also serves as a competitive inhibitor limiting PKR binding to eIF2α thanks to a sequence homology between one of its motifs and eIF2α [88,89,90].
Cells experiencing ER stress due to nutrient deprivation or viral replication may lead to the activation of PERK. Like many other viruses, HIV-1 is known to induce protein misfolding and ER stress [91]. Campestrini et al. have previously reported on the induction of ER unfolded protein response (UPR) genes such as PERK and ATF4, following the treatment of Jurkat cells with the HIV-1 Tat protein [11]. Additionally, PERK activation has been observed in Tat-treated human brain microvascular endothelial cells (HBMECs) and in HIV-1-infected CD4+ T cell lines [92,93]. While PERK is one of the sensors of the UPR with IRE-1 and ATF6, the functional role of PERK in ATF4 induction in response to HIV-1 infection is not fully demonstrated.
GCN2 is activated in response to amino acid deprival and metabolic dysfunction (Figure 1). These alterations are reported in plasma of HIV-1-infected patients and in the gut of SIV-infected monkeys [8,94,95,96,97]. Moreover, GCN2 is activated in vitro by HIV-1 or SIV infection [8,98] suggesting that metabolic disorders observed in HIV-1-infected patients could also lead to GCN2 activation. GCN2 phosphorylates the HIV-1 integrase, which reduces viral integration [99]. This property is not limited to HIV-1 since various integrases from other retroviruses are also recognized as substrates by GCN2 [99]. The anti-viral activity of GCN2 can be overcome by the HIV-1 protease that can cleave GCN2 [100]. Jiang et al. have reported that HIV-1-induced ATF4 transcription would result from the activation of the GCN2/ATF4 pathway [8]. Serum deprivation triggers HIV-1 replication of CD4+ T cells, correlating with the GCN2-mediated activation of ATF4, which is recruited to the HIV-1 long terminal repeat (LTR) to facilitate viral transcription.
Both heme/iron depletion and arsenite-induced oxidative stress activate HRI [3]. Arsenite-induced HRI activation increases viral protein production and the infection rate of reovirus [101]. It can also regulate the level of the viral factor Zta that plays a role in the lytic cycle of EBV [102,103]. HRI has been shown to be able to activate ATF4, triggering the expression of genes involved in autophagy [104,105,106] and oxidative-stress response including the antioxidant heme oxygenase-1 (HO-1) gene [106]. Different groups have reported a role of HO-1 in regulating viral replication [107,108] including HIV-1′s [109]. HO-1 dysfunction has been associated with neuronal diseases in people living with HIV-1 [110]. For instance, despite a link between HO-1 and viral replication, to date, no study has addressed the role of an HRI pathway regulating HIV-1 infection and replication.
Recently, two other kinases able to phosphorylate eIF2α have been characterized, namely MARK2 and FAM69C [70,71]. Both kinases are induced by proteotoxic stress. FAM69C KO mice-derived microglia displayed an indirect increase in inflammation, and HIV-1 binds MARK2 to control motor adaptor function on viral cores [71,111]. However, no HIV-1-induced ATF4 activation depending on these kinases has been reported yet.
To conclude, PERK and GCN2 may lead to ATF4 activation after HIV-1 infection, and the direct interaction of HIV-1 components with GCN2 and MARK2 may in turn control their ability to activate ATF4. The potential control of ATF4 by the new eIF2α kinases needs further investigation during HIV-1 infection.
2.2.2. Mitochondrial Stress Response, ATF4 and HIV-1
The contribution of HIV-1 infection to the induction of mitochondrial dysfunctions has been described earlier [112,113,114]. During HIV-1 infection, mitochondrial functions are compromised resulting in reduced oxidative phosphorylation (OXPHOS), ATP synthesis, gluconeogenesis, and β-oxidation. In addition to membrane depolarization and release of cytochrome C [115,116], HIV-1-induced alterations may also disrupt cellular homeostasis, increase oxidative stress, affect mitochondrial dynamics, and lead to the loss of mitochondrial DNA [115,117,118]. Mitochondrial dysfunctions are not only observed in CD4+ T cells, but also in myeloid cells such as neutrophils [119,120], monocytes [121], and CD8+ T cells [122], which are non-infected T cells. Therefore, indirect mechanisms may contribute to the alteration of mitochondrial functions during HIV-1 and SIV infections.
ATF4 is induced by several mitochondrial stress-like alterations affecting proteostasis, respiration, and mitochondrial membrane potential (MMP) loss [123,124,125]. A multi-omics study has suggested that ATF4 coordinates the mitochondrial stress response [125]. ATF4 induction was shown to depend on HRI in response to the respiratory chain and ATP synthesis disruptions [126,127]. Individual knockdown of each of the four original ISR kinases (i.e., HRI, PERK, PKR and GCN2) did not abolish the induction of ATF4 and its target genes. These results could suggest the role of either unknown or newly identified ISR kinases (as indicated above), or some redundancy.
In response to mitochondrial misfolded protein accumulation, ATF4 plays a significant role in cell homeostasis as a transcription factor of the canonical mitochondrial UPR (UPRmt) [124]. ATF4 is also associated with the transcription of genes encoding mitochondrial chaperonin and proteases [124]. For instance, Li et al. suggest that unfolded mitochondrial proteins would be degraded by lysosomes, leading to the increase in amino acids that would activate mTORC1 via lysosomal v-ATPase through a still-unknown mechanism during the UPRmt [128,129,130,131]. mTORC1 would then phosphorylate and activate ATF4, thereby triggering transcription of chaperone-encoding genes, and increasing the mitochondrial folding capacity [129].
ATF4 is also activated in response to alterations in mitochondrial dynamics. The deletion of optic atrophy protein 1 (OPA1), which is crucial for the fusion of inner membranes during mitochondrial fusion, and contributes to the release of apoptogenic factors [132,133], generates mitochondria-derived reactive oxygen species (ROS) and thus causes increased oxidative stress and death [117]. This process triggers ER stress and a PERK-dependent UPR, resulting in the transcriptional activation of ATF4 and other genes. This response initiates a catabolic program contributing to muscle loss and systemic aging [134]. The knockdown of Drp1, a major effector of mitochondria fission [135], leads to eIF2α phosphorylation and ATF4 activation in the liver [136]. Although modulation of OPA1 was not directly associated with ATF4 activation, it has been show that the OMA1 protease, which cleaves the mitochondrial protein DELE1, but also OPA1 [137,138], leads to the release in the cytosol of the DELE1′s carboxy-terminal domain that oligomerizes with HRI (Figure 1) [126,127,139].
2.2.3. The Viral Vpu Protein Stabilizes the ATF4 Protein
The HIV-1 viral protein U (Vpu) is an HIV-1 accessory protein that down-modulates CD4 and BST-2/tetherin. A cellular Skp, Cullin, F-Box (SCF) E3 ubiquitin ligase complex is recruited by Vpu to target CD4 for ubiquitination and proteasomal degradation [140]. This process involves the recruitment of the F-box β-transducin repeat-containing protein (βTrCP) [141,142,143,144]. Additionally, Vpu reduces BST-2/tetherin from the cell surface by preventing the trafficking of BST-2/tetherin to the plasma membrane from the trans Golgi network and/or the recycling endosome [145,146,147]. Furthermore, Vpu targets BST-2/tetherin for degradation, thus promoting viral progeny release and inhibiting NF-κB signaling [143,148].
The SCF-βTrCP E3 ubiquitin ligase complex has been reported to contribute in the degradation of ATF4 (Figure 1) [149]. Unlike its effect on CD4 and BST-2/tetherin, Vpu inhibits ATF4 βTrCP-mediated proteasomal degradation [150]. This apparent discrepancy in the effect of Vpu on βTrCP-dependent proteasomal degradation may be explained by the existence of two distinct paralogs of β-TrCP, βTrCP1/BTRC and βTrCP2/FBXW11 [151]. Recent work by Pickering et al. demonstrates that Vpu has contrasting effects on βTrCP1 and βTrCP2 and suggests that Vpu would induce proteasomal degradation mediated by βTrCP2 and inhibit βTrCP1-dependent protein degradation [152]. Thus, the contribution of viral proteins encoded by HIV-1 merits further investigation regarding the role of ATF4.
2.2.4. HIV-1 Antiretroviral Drugs Induce ATF4 Signaling
HIV antiretroviral therapy (ART) has drastically altered the course of HIV-1 infection, resulting in a major decrease in morbidity and mortality. However, drug side effects have been reported earlier, leading to their progressive replacements and the development of new molecules. Thus, mitochondrial damage was initially reported following the use of reverse transcriptase inhibitors (RTIs) and protease inhibitors (PIs) [153,154,155,156,157]. In addition to mitochondrial stress, it has been shown that Nelfinavir (PI) triggers an ATF4 transcriptional response associated with liver metabolic alterations that have been reported in PLWH [158] and causes cell cytotoxicity against ovarian cancer cells [159,160]. In addition to Nelfinavir, it has been shown that Lopinavir (PI) also increases the level of ATF4 transcript in SQ20B and FaDu cancer cell lines [161] but in a model-dependent manner because no effect was observed on the level of ATF4 transcript in a model of trophoblast cell differentiation [162]. While Zidovudine (AZT, RTI), which induces mitochondrial stress, was recently reported to extend the lifespan of C. elegans depending on ATF4 activation [163], a reduction in ATF4 expression was reported [164] in long-term Tenofovir disoproxil fumarate (TDF)-treated individuals presenting a decrease in bone mineral density. Therefore, the role of ATF4 remains to be clarified regarding the use of RTIs. Interestingly, an HIV-1 integrase inhibitor (IN), namely MK-2048, was shown to selectively kill HTLV-1–infected cells by inducing the PERK/ATF4/CHOP pathway [165]. This molecule could be also of interest for people living with HIV-1 (PLWH), in which triggering the death of viral infected cells may reduce the extent of viral reservoirs. Thus, several ARTs may provide a beneficial effect not only by tackling HIV viral replication but also by stimulating ER stress via ATF4 and thus facilitating the death of infected cells.
3. ATF4 Role during HIV-1 Replication
3.1. ATF4 Positively Regulates HIV-1 Cycle
The induction of ATF4 during HIV-1 infection raises the possibility that ATF4 may play a role in viral replication. Thus, it has been shown that the overexpression of ATF4 promotes viral replication, whereas its silencing suppresses HIV-1 replication [6,7]. Using compounds that inhibit GCN2 or PERK or that lead to increases in ATF4 levels, it has been shown that the induction of the ISR/ATF4 pathway reactivates HIV-1 in models of HIV-1 latency [8,9,10]. Altogether, these data strongly suggest a role for ATF4 in regulating HIV-1 replication both during the acute infection and the exit from latency. However, given the nature of infected cells that include memory CD4 T cells and T follicular helper cells (Tfh) [166,167,168,169,170], which represent the main reservoirs in visceral tissues, further analyses should be performed to elucidate the role of ATF4 in primary T cell subsets.
3.2. How ATF4 Favorizes HIV-1 Replication
3.2.1. ATF4 Binds to the HIV-1 LTR and Promotes Viral Gene Transcription
ATF4 can be a direct regulator of HIV-1 transcription. During HIV-1 infection, various cellular transcription factors including some bZIP domain proteins have been shown to bind the 5′ long terminal repeat (LTR)—that contains the transcriptional promoter of the viral genome of HIV-1—and regulates transcription of HIV-1 genes [171] (Figure 2).
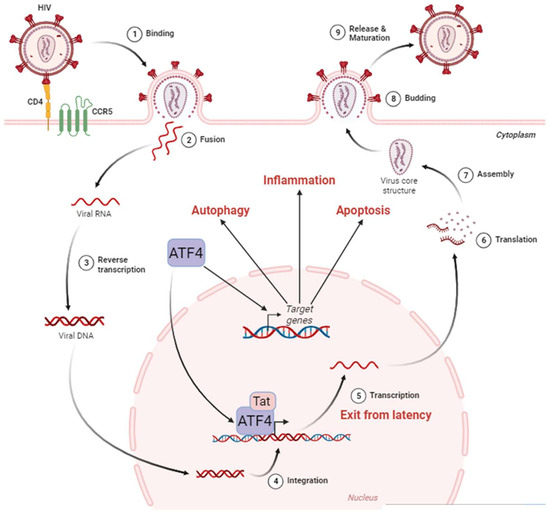
Figure 2. ATF4 in the HIV-1 cycle. The HIV-1 cycle begins with the attachment of the virus to the cell surface thanks to the CD4 and CCR5 receptors (1). This allows the fusion between the viral envelope and the host cell membrane (2). During this phase, the viral nucleocapsid enters the cell cytoplasm. Single-stranded RNA molecules are then retro-transcribed in the nucleus (3). The viral DNA then integrates into the host cell genome (4), where it can remain latent until reactivation. The exit from the latency is stimulated by ATF4, which, in cooperation with the viral protein Tat, activates the HIV-1 promoter located in the LTR region of the viral DNA leading to viral RNA (5). These HIV-1 messenger RNAs are exported from the nucleus to be translated (6) into proteins that assemble to form the internal structure of viral particles (7). Budding from the cell membrane (8) culminates with the release and maturation of viral particles in the extracellular environment (9). ATF4 is also capable of inducing the activation of target genes involved in cellular processes such as autophagy, inflammation, and apoptosis. Adapted from “Disease Mechanism–Infectious Diseases, HIV Replication Cycle”, by BioRender.com. Retrieved from https://app.biorender.com/biorender-templates (accessed on 18 January 2024).
ATF4 induces HIV-1’s gene transcription and regulates the Human T-cell Leukemia Virus (HTLV) promoter [172]. As an ATF/CREB family member, ATF4 binds to the C/EBP-ATF consensus sequence (TGACGT (C/A) (G/A)). Two C/EBP-ATF-binding sites were first identified in the LTR of HIV-1, but super-electrophoretic mobility shift assay (EMSAs) failed to show the binding of ATF4 to these regions [173,174]. A bioinformatics approach led by Jiang et al. identified additional potential C/EBP-ATF-binding sites in the LTR [8]. Caselli et al. reported that co-expression of Tat and ATF4 induced higher LTR transcription than the sole expression of Tat, suggesting a synergistic interaction between the two proteins [6]. ChIP experiments showed that ATF4 binds to the LTR sequence. This interaction is positively regulated by Forkhead box O 1 (FOXO1) inhibition and amino acids deprival [8,9]. Further works suggest that Tat may be unnecessary for ATF4-mediated HIV-1 promoter activation [6,7]. However, this effect has only been observed in HeLa cells, but not in Jurkat and 293 T cells [6].
ATF4 can also bind to sequences that differ from the C/EBP-ATF consensus sequence, depending on its dimerization partners (Figure 3) [1,175,176]. ATF4 can dimerize with Jun and Fos [177] that heterodimerize to form the AP-1 transcription factor [177]. The fact that the HIV-1 promoter presents an AP-1-binding site could suggest that a heterodimer of ATF4 with Jun or Fos can be formed to activate the LTR activity. However, the partners able to dimerize with ATF4 to control HIV-1 transcription are still unknown.
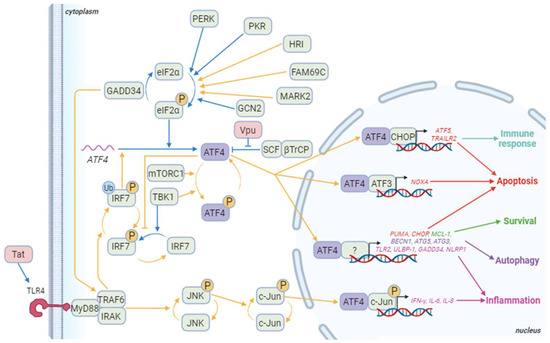
Figure 3. ATF4 signaling pathways during HIV-1 infection. Blue arrows correspond to interactions demonstrated in HIV-1 literature. Yellow arrows correspond to other contexts. Besides ATF4, cellular proteins appear in green and HIV-1 proteins in red. The genes in red are some of the pro-apoptotic genes induced by ATF4/ATF3 or ATF4/CHOP dimers and ATF4 dimerizing with a partner that remains to be determined. ATF5 also regulates the immune response at least by controlling immune cell differentiation. ATF4 also controls the expression of several pro-apoptotic genes including TID1/DNAJA3, G0S2, and TP53BP2. The genes in green are involved in ATF4-mediated survival as TMBIM5. Genes in purple are implicated in ATF4-induced autophagy like WIPI1, ATG12, ATG10, ATG16L1, ATG7, MAP1lc3B, GABARAPl2, p62/SQSTM1, NBR1, and REDD1. Finally, genes in pink are genes related to inflammation that are induced by ATF4 in a dimer with phosphorylated c-Jun or an undefined partner. Other genes activated by the phosphorylated ATF4/c-Jun dimer include RANTES and sICAM-1. Not shown in this diagram is IRF7 activation, which induces the production of type I and II interferons, and then activates the PKR/eIF2α/ATF4 pathway. Created with BioRender.com (accessed on the 18 January 2024).
Five to twenty percent of HIV-1-infected patients are co-infected by the Hepatitis B virus (HBV). The X protein of HBV has been shown to regulate HIV-1 transcription by stimulating the binding of ATF4 to the LTR of HIV-1 [178]. ATF4 can also directly interact with the HTLV-1 transcriptional activator Tax protein and act as an adapter between Tax and the HTLV-1 promoter [172,179]. Therefore, co-infections may provide a permissive environment for ATF4 to activate the HIV-1 promoter.
3.2.2. ATF4, HIV-1 and Apoptosis
It is well known that HIV-1 pathogenicity is associated with the depletion of CD4+ T cells leading to the development of AIDS. In this context, it has been shown that a form of programmed cell death, namely apoptosis, may contribute to the depletion of CD4+ T cells and be associated with pathogenicity [65,180,181]. Both in vitro and in vivo, HIV-1 and SIV infections mediate T cell death [67,182] through the intrinsic apoptotic pathway that is characterized by the expression of the pro-apoptotic proteins Bax, Bak, and Bim [183], leading to the release of apoptogenic mitochondrial factors and consequently to caspase activation. CD4+ T cell apoptosis also involves the extrinsic pathway in which Fas (CD95) is critical [184,185,186,187]. Thus, caspase inhibition prevents in vivo the depletion of CD4+ T cells and delays the progression to AIDS [66].
It has been shown that Tat up-regulates UPR mediators with an increase in the transcript levels of ATF4, CHOP and the pro-apoptotic BH3-only BIM (BCL2L11), thus leading to apoptosis [11]. Of interest, ATF4 mediates cell death through the activation of the bZIP transcription factor CHOP [3], which also induces the transcription of BIM and of the pro-apoptotic BH3-only PUMA (also known as BBC3) [188,189]. Additionally, the CHOP-ATF4 dimer up-regulates ATF5, NOXA (also known as PMAIP1), APAF-1, and TXNIP, which in turn amplify cell death [190,191,192]. ATF4 also promotes degradation of the caspase inhibitor X chromosome-linked inhibitor of apoptosis (XIAP) protein through the ubiquitin-proteasome system, which allows the activation of caspases [193].
Altogether, the identified ATF4 targets represent multiple new mechanisms to be explored by which ATF4 could regulate cell death or survival of HIV-1-infected cells.
3.2.3. ATF4, HIV-1 and Autophagy
Autophagy is a pro-survival intracellular catabolic process that targets protein aggregates and damaged organelles in response to stress. ATF4 activates the transcription of genes involved in the initiation (BECN1), in the elongation of the phagophore and the maturation of the autophagosome (WIPI1, ATG12, ATG5, ATG10, ATG16L1, ATG3, ATG7, MAP1lc3B, GABARAPL2), and in the selective clearance of cargo (p62/SQSTM1 and NBR1) [175,236]. Interestingly, ATF4 triggers the transcriptional activation of the autophagy genes directly as a homo or heterodimer with CHOP or indirectly through CHOP induction (Figure 3) [236]. The ratio between ATF4 and CHOP is decisive for the regulation of these genes, underpinning a fine-tuned control of the different stages of autophagy, the subtleties of which remain to be elucidated. ATF4 can also act upstream of autophagy to induce this process; ATF4 can increase the expression of the gene encoding Regulated in development and DNA damage response 1 (REDD1; also known as Ddit4), which suppresses mTOR complex 1 (mTORC1) activity, allowing autophagy induction upon ER stress and starvation [237,238,239].
HIV-1 modulates autophagy in a cell-type-dependent manner [240,241]. The Env protein found at the cell surface of infected cells induces an autophagy-dependent cell death of bystander CD4+ T cells [242]. Thus, ARF6, which promotes autophagosome biogenesis by facilitating endocytic uptake of the plasma membrane into autophagosome precursors, has been proposed to promote the fusion of HIV-1 envelope with the plasma membrane, therefore permitting the entry of HIV-1 in CD4+ T lymphocytes [243,244]. Other groups have reported that HIV-1 inhibits autophagy [245,246]. After virus entry, the Vpr protein decreases the amount of proteins like MAP1LC3 and BECN1 [247]. Furthermore, although autophagy is initiated in primary CD4+ T cells infected with HIV-1, this process aborts due to a lysosome destabilization by DRAM, a p53-inducible gene. This permeabilization of lysosomes and resulting cell death has been proposed as an altruistic self-defense mechanism limiting viral dissemination by the elimination of infected cells [220]. Despite the obvious link between ATF4 and autophagy, the role of ATF4 in autophagy in the context of HIV-1 infection remains poorly documented.
3.2.4. Immune Response and ATF4 Activation during HIV-1 Infection
ATF4, innate immunity and HIV-1
Pattern recognition receptors (PRRs) are fundamental molecules for innate immune response recognizing pathogen-associated molecular patterns (PAMPs). PRRs bind to PAMPs and trigger cell signaling cascades. PRRs include Toll-like receptors (TLRs), Nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), RIG-I-like receptors (RLRs), and C-type lectin receptors (CLRs). For example, TLR7 and TLR8 can recognize single-stranded RNAs, which are present in the HIV-1 genome [248], leading to Interferon regulatory factor 3 and 7 (IRF3 and IRF7) activations and inducing robust expression of type I interferon (IFN) genes [249]. ATF4 inhibits IRF7 transcription but also its activation by inhibiting TANK-binding kinase 1 (TBK1) and IκB kinase (IKK)ε-mediated IRF7 phosphorylation [57]. To be activated, IRF7 also requires its ubiquitination by Tumor necrosis factor receptor-associated factor 6 (TRAF6) (Figure 3). GADD34 that binds to and inhibits TRAF6 activity is an ATF4 target [250]. Its transcript levels have been shown to be increased in HIV-1-infected cells in a p53-dependent manner [251]. Inhibition of TRAF6 enhanced HIV-1 replication in macrophages [252], whereas GADD34 reduced viral protein expression [253]. Therefore, the role of ATF4 and its targets merits to be further explored in innate immunity.
ATF4, cellular immunity and HIV-1
Viruses have developed a diverse array of strategies to manipulate host cell metabolism and reorientate metabolic resources toward their benefit [118]. Current knowledge on T-cell metabolism in HIV-1 infection suggests that HIV-1 takes advantage of the glycolytic process in CD4+ T cells to infect them and to boost viral replication [261,262,263]. It has been shown that IL-7-mediated thymocytes survival through the increase in Glut1 protein levels at the cell surface, leading to glucose uptake and favoring viral infection [264]. Interestingly, ATF4 can influence a network of genes responsible for metabolic flux when activated by an extracellular oxidizing environment [265]. Thus, ATF4 up-regulates genes involved in glycolysis and promotes the anaplerotic flux through enhanced glutaminolysis, which plays a role in T cell growth in oxygen- or amino-acid-deprived environments [265]. Glutaminolysis fuels the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) in T-cell receptor-stimulated naïve CD4 subsets, as well as memory CD4 subsets [266]. This balance in the bioenergetics pathways is essential for T cell polarization [267]. ATF4 has also been implicated in regulating the differentiation of naïve T cells in T helper 17 cells (Th17). T cells from ATF4-deficient mice are characterized by a diminished Th1 differentiation and an elevation in factors promoting Th17 commutation, such as Interleukin-17 (IL-17) [265]. This association is further supported by the effect of the small molecule halofuginone (HF), which inhibits Th17 differentiation in both mice and humans and increases ATF4 protein levels [268]. Notably, adding amino acids in excess counteracts the inhibition of Th17 differentiation by HF, suggesting the involvement of the amino acid starvation response mediated by ATF4 in targeting Th17 differentiation. Interestingly, Th17 cells are highly vulnerable to HIV-1 infection [269], and this population represents a major viral reservoir under ART [270]. The loss of Th17 homeostasis contributes to disease progression in SIV-infected rhesus macaques associated with the loss of intestinal epithelial integrity, leading to microbial translocation [271,272,273,274]. This alteration in the balance of T cell subsets persists despite ART [275]. Given the role of ATF4 in regulating mitochondrial and reticulum stress-associated apoptosis, a contribution of ATF4 dysregulation in the alteration of the Th17 balance at the mucosal barrier cannot be excluded. Taken together, these results suggest that the induction of ATF4 observed in CD4+ T lymphocytes during HIV-1 infection [10] may contribute to the increased production of energy and amino acids required for viral replication.
This entry is adapted from the peer-reviewed paper 10.3390/biology13030146
This entry is offline, you can click here to edit this entry!