Molecular and cellular research modalities for the study of liver pathologies have been tremendously improved over the recent decades. Advanced technologies offer novel opportunities to establish cell isolation techniques with excellent purity, paving the path for 2D and 3D microscopy and high-throughput assays (e.g., bulk or single-cell RNA sequencing). The use of stem cell and organoid research will help to decipher the pathophysiology of liver diseases and the interaction between various parenchymal and non-parenchymal liver cells. Furthermore, sophisticated animal models of liver disease allow for the in vivo assessment of fibrogenesis, portal hypertension and hepatocellular carcinoma (HCC) and for the preclinical testing of therapeutic strategies. The purpose of this review is to portray in detail novel in vitro and in vivo methods for the study of liver cell biology that had been presented at the workshop of the 8th meeting of the European Club for Liver Cell Biology (ECLCB-8) in October of 2018 in Bonn, Germany.
- hepatic stellate cells
- hepatocellular cancer
- fibrogenesis
- steatosis
- in vitro models
Introduction
Cell culture techniques are important tools for the study of pathogenesis and treatment of liver diseases. Each different cell type plays a specific role in the liver. The parenchymal cells, mainly hepatocytes, constitute 80% of the total liver volume and are responsible for the majority of liver functions. The nonparenchymal cells include hepatic stellate cells (HSCs), liver sinusoidal endothelial cells and resident macrophages such as Kupffer cells (KCs) [1]. The connective tissue of the liver consists of blood vessels, nerves, lymphatic vessels, bile canaliculi and extracellular matrix [2]. Inside the bile canaliculi, the epithelial cells, the so-called cholangiocytes, participate in the regulation of bile production and in the process of biliary repair [3]. In recent years, biliary research and cholangiocytes gained much interest in the research of liver diseases. Although conventional procedures to isolate liver cells, such as in situ liver perfusion and collagenase digestion followed by density gradient isolation, are well established, novel methods have been introduced such as fluorescence-activated cell sorting (FACS) of HSCs using vitamin A autofluorescence. These approaches provide high purity of HSCs and are reproducible. In the coming years, the use of 2D microscopy, 3D microscopy, intravital microscopy, flow cytometry and cell isolation for subsequent functional experiments or expression analyses is expected to significantly increase characterization of hepatic cell populations and their interactions with circulating immune cells. For instance, these methods have been used to study the impact of hepatic macrophages and monocytes on steatosis, inflammation, hepatocellular injury, HSCs activation and angiogenesis [4]. Two novel technologies from stem cell research, induced pluripotent stem cells (iPSCs) and liver organoids, helped to decipher the pathophysiology of cholangiopathies [5]. Using in vitro models of primary human liver cells from different human donors, co-culture systems can simulate additional pathophysiological liver cell interactions, being complementary to animal models in preclinical analysis. Furthermore, development of animal models to experimentally mimic alcoholic liver disease (ALD), such as the liquid Lieber–DeCarli (LDC) diet, intragastric ethanol infusion and administration of carbon tetrachloride (CCl4) combined with ethanol in drinking water, can help to understand critical pathophysiological steps of human ALD, such as inflammation and fibrosis during ALD progression [6][7]. Moreover, HCC models are receiving increased attention, not only for the fast xenograft model, but also for application of N-nitrosodiethylamine (DEN) followed by repeated administration of CCl4 (Table 1) [8], as well as the model of non-alcoholic steatohepatitis (NASH) combined with a HCC model described recently by using a Western diet (WD) combined with CCl4 (Table 1) [7][9]. Finally, the assessment of systemic, splanchnic and portal hemodynamics in animal models is now well accepted and crucial for the evaluation of drugs for end-stage liver disease. The purpose of this review is to present technological tools that will enhance our ability to isolate and purify the different cell populations and other strategies to study the full spectrum of liver disease (Figure 1).
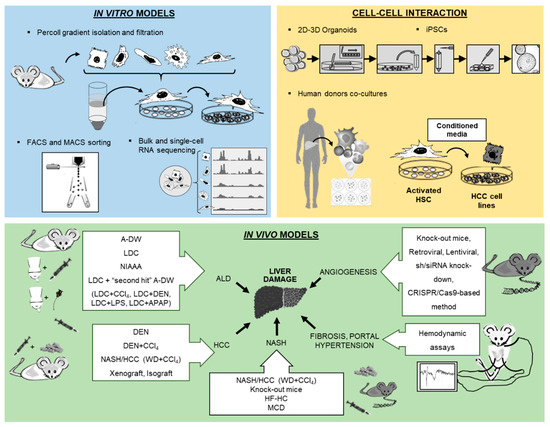
Figure 1. Overview of new methodologies using in vitro, cell–cell interaction and in vivo models for the study of liver pathology.
Table 1. Select findings of new methodology applications in in vitro organoids and in vivo models.
In Vitro Models
Isolation and Culture of Hepatocytes
Hepatocytes are the major cell population in the liver [57]. Hepatocytes are responsible for management of nutrient uptake, blood detoxification and packaging and secretion of proteins, lipids and bile [58]. Their isolation was well established by the two-step EGTA/collagenase perfusion technique by Seglen in 1976 [59]. The 2D culture of primary hepatocytes is considered the “gold standard” for in vitro testing of the hepatotoxic effect of therapies and drug metabolism [60]. The disadvantage of 2D culture is that within a few days, hepatocytes suffer dedifferentiation and loss of function. The 3D techniques are gaining relevance due to the presence of the non-parenchymal cells that play the role of maintaining hepatocyte functions [28]. Developments in the production of 3D hepatocyte culture scaffolds are helpful in understanding the cross talk between hepatocytes and non-parenchymal cells and their role in the regulation of liver pathologies.
Cellular stress during the isolation techniques results in a low quality of isolated cells and reduced cell engraftment after transplantation. The use of 3D cell formats has also been successful in processes of cryopreservation and post-thawing, improving in vivo efficacy [28]. Furthermore, applying good manufacturing practices (GMPs) improves hepatic functions and the ability to engraft in vivo, compared to the classic organ storage solution based on the University of Wisconsin medium [61].
Hepatocytes-like cells derived from subsequent differentiation of iPSCs using a cocktail of growth factors and specific matrices is another alternative technique to obtain hepatocytes in vitro [24][25][62]. One disadvantage is that iPSC-derived hepatic cells are phenotypically more similar to fetal hepatocytes than to the freshly isolated counterpart [63]. To overcome this problem, new strategies include a stepwise induction with cocktails of small molecules to improve the final maturation [26][64].
HSCs Isolation and Immortalized HSCs Lines
HSCs, the main vitamin A-storing cells located in the perisinusoidal space between hepatocytes and sinusoids, play a key role in collagen production, secretion and function of cytokines and chemokines, as well as in the modulation of the immune system in addition to changes in contractile features during homeostasis and liver fibrosis [65]. It has now become clear that not all functions are done by the same HSC, but that the transcriptional profile of single HSCs considerably varies after activation in vivo, particularly during fibrogenesis [4]. First protocols for HSC isolation were established in the 1980s, with collagenase/pronase digestion of the liver tissue and subsequent fractioning process of the heterogeneous cell suspension on a density gradient [66]. In 1998, other methods to isolate HSCs appeared, such as the first protocol to isolate HSCs with FACS. This involved applying FACS to sort cells from rats with high purity, using simply their high side scatter of incident light (Table 1) [10]. Later, FACS instruments equipped with a UV laser could sort HSCs by visualizing the typical autofluorescence from vitamin A storage (Table 1) [11][12].
HSC isolation with FACS sorting allows isolation with high purity, and, compared to standard techniques, FACS-based protocols can be used to isolate HSCs from much younger animals, such as genetically modified mice characterized by a short life span. Moreover, these protocols improve isolation of HSCs when hepatocytes are fattened in steatosis models. In addition, these protocols permit simultaneous comparison of hepatic cell subpopulations from the same animal. Despite these advantages, FACS sorting requires investment in special equipment, including UV lasers, appropriate filters and specific skill acquisition. Furthermore, these protocols require pooling of livers, long time periods for cell sorting and careful use of UV light, which can be stressful and cause cell damage.
In summary, HSC sorting by FACS via vitamin A autofluorescence provides the opportunity to obtain excellent cell purity and, despite the fact that isolated HSCs cannot reproduce the same phenotype as the one found in cirrhotic livers, these protocols can be used to optimize the study of HSCs biology and their role in liver fibrosis. Undeniably, the co-culture systems of HSCs with other cells could explain the relevant role of cell–cell interactions and the paracrine influences. This has been exemplarily demonstrated more than a decade ago for the co-culture of HSC with Kupffer cells that mimicked the in vivo activation of HSC much more accurately than single HSC cultures [67].
Analysis of Liver Cells from Different Human Donors and Co-Culture Models
Despite the emergence of therapeutic targets for liver pathologies, the response of the treatments shows significant variations between patients. In vitro studies with liver cells from different human donors could help to understand and determine the differences observed in in vivo studies. Use of complex co-culture models could be a start to better simulate the complexity of the in vivo situation. This is currently intensively explored for fatty liver disease conditions, in which novel 3D biochip systems partially allow modelling complex cellular interactions between steatotic hepatocytes and non-parenchymal cells [68]. However, for the comparison of primary liver cells from different human donors, it is critical to exclude that isolation procedures can affect functional cellular characteristics. Therefore, it is important that it has been shown that different HSC isolation procedures from tissue of the same donor resulted in no significant differences, whereas HSCs isolated from different human donors revealed significant variations (Table 1) [16].The differences in the expression levels of profibrogenic genes observed between HSCs from different human donors [69] may reveal some aspects of varying fibrosis progression in patients as well as development of HCC.
Furthermore, numerous studies have shown that HSCs are related to the formation and progression of HCC (Table 1) [17][18]. The treatment of human HCC cells with conditioned media of HSCs from different human donors resulted in significantly different functional effects as well as gene expression changes in the HCC cells [70]. Bioinformatic modeling led to the identification of pregnancy-associated plasma protein A (PAPPA) as novel cancer-promoting stromal factor secreted by HSCs, which is related to advanced-stage HCC [70]. In addition, HSCs can also be used in co-culture models with other cells, such as HSCs treated with conditioned media from different human melanoma cells (Table 1) [19], or conditioned media from steatotic human hepatocytes that produce a more fibrotic phenotype in HSCs compared with normal hepatocytes (Table 1) [20].
In summary, in vitro (co-)culture models of primary human liver cells from different human donors can be a valuable system to simulate at least certain aspects of the heterogeneity and variation of the course of chronic liver disease. Furthermore, in vitro studies with human liver cells may be used to predict the response of patients prior to defined therapies.
Isolation and Characterization of Liver Macrophages
Liver macrophages play a key role in innate immunity, homeostasis and inflammation. Among liver macrophages, two principal populations can be differentiated by their ontogeny, polarization and function during injury and resolution [71], namely KCs and monocytes, and their functional behavior in liver diseases could become a target for novel therapeutics (Table 1) [38]. Liver macrophages have been characterized by means of new technologies, such as multicolor flow cytometry, advanced microscopy, sorting followed by bulk sequencing or single-cell RNA sequencing (Table 1) [4][13][21][22][23][72]. From an isolation perspective, KCs are firmly attached to sinusoidal endothelial cells and require gentle perfusion-based dissociation methods [73], while monocytes can be isolated simply by collagenase digestion (Table 1) [40]. These protocols are based on perfusion of the liver via portal vein cannulation, cell extraction, purification via density gradient or sedimentation and optional additional steps for cell sorting based on FACS or MACS. The inconvenience of these techniques lies in the activation of hepatic macrophages during isolation and the fact that they also require adaptation based on the mouse strains used, making cross-lab standardization difficult. Moreover, compared to the MACS sorting method, FACS usually results in excellent purity and can be flexibly adjusted to the populations of interest. However, FACS can impact cell viability, it is time-consuming and expensive. Similar to HSC, macrophages behave differently in vitro, if cultured alone, together with other cells or in the context of the liver structure with zonation, flow conditions and nutrient / oxygen gradients.
Many studies are currently performed to understand human liver macrophages. Initial reports using single-cell RNA sequencing confirmed the heterogeneity and functional diversification of macrophages in healthy and diseased human livers [23][72]. However, important pitfalls include the limited availability of human samples, ischemic alteration of tissue during surgical procedures, heterogeneity of patients and the relative paucity of specific markers for hepatic macrophage subsets in humans [71]. For instance, the c-type lectin “clec4f” is considered specific for KCs in mice, but it has no direct counterpart in humans [74][75].
Cellular Models in Biliary Research
Cholangiocytes are epithelial cells of the bile ducts. Their function is to modify the composition and volume of bile produced by hepatocytes en route to the duodenum. Different sub-populations of cholangiocytes exist: the large cholangiocytes involved in secretory processes and the smaller cholangiocytes equipped with plasticity to proliferate in response to damage [76]. Cholangiopathies represent a significant cause of liver-related morbidity and mortality and are an important indication for liver transplantation [77].
Cholangiocyte isolation is realized firstly by in situ liver perfusion and collagenase digestion, secondly by mechanical and enzymatic digestion, and finally the cells are separated by filtration. At this point, it is possible to obtain different sizes of cells, intrahepatic bile duct units (IBDUs) (Table 1) [14] or, via immune-magnetic separation, biliary epithelial cells (BECs) (Table 1) [15]. While IBDU isolation is not completely pure, it is sufficient for secretory function studies 48 h after plating on matrigel [78][79][80]. In contrast, BEC can be more purified but cannot proliferate in culture. Recently, it has been shown that primary cultures of mouse cholangiocytes could be cultured on rat tail collagen for several passages [81] and used for specific functional studies with transgenic mice [82].
Cholangiocytes from human liver explant tissue can also be isolated by collagenase digestion followed by Percoll gradient isolation and immune magnetic positive selection and can then be expanded in culture since they acquire a more mesenchymal phenotype [83]. Furthermore, it is possible to culture cholangiocytes from a small fragment of a liver biopsy, but with a lower purification [84]. Surprisingly, these cholangiocyte phenotypic markers are established: γ-glutamyl-transpeptidase (GGT), cytokeratins 7 and 19, EpCam, SOX-9, secretin receptor and cystic fibrosis transmembrane conductance regulator (CFTR) [85].
The study of human cholangiocytes is relevant to the understanding of the biological functions of the biliary epithelium and cholangiopathies. Despite relentless efforts to isolate cholangiocytes, the main challenge lies with the yield and the purity of the cell preparation technique since these represent about 4% of the total liver cell populations, similar in size to endothelial and KCs. Other limitations are the need of a polarized organization to improve their functionality, the time-consuming method and the complex media needed for expansion.
Compared to other liver cell isolation models, accessibility to human tissue is limited and restricted to the end stage of liver disease. In the case of animal models, there are physiological inter-species differences. A number of other options are currently under investigation, such as iPSCs and the possibility to isolate 3D liver organoids, which may provide opportunities to overcome current limitations (Table 1) [27]
In Vivo Models
Angiogenesis and Gene Expression Inhibition
One of the hallmarks of chronic and liver disease is angiogenesis with new formation of blood vessels from preexisting vasculature [29][86]. In this pathological condition, abnormal angioarchitecture is established together with fibrogenesis, inflammation and tumorigenesis, resulting in the formation of portosystemic collateral vessels, an increase in splanchnic blood flow and the aggravation of portal hypertension. All of these could be targets for new therapeutic approaches in treating angiogenesis in chronic liver disease (Table 1) [29][86].
To address angiogenesis, a number of different methods have been developed in recent decades [87]. Using in vitro bioassays, purified endothelial cell cultures or co-cultures with supporting cells (e.g., smooth muscle cells, pericytes, fibroblasts and tumor cells), either on 2D monolayers or 3D spheroids, were employed to mimic aspects of in vivo vascular formation. However, there are some critical points to consider regarding the in vitro assays and the choice of endothelial cells. Different factors characterize endothelial cells: they have species- and organ-associated phenotypic differences and depend on their microvascular origin. Also, the number of passages, cells plated per well and the use of growth factor-reduced matrigel are factors influencing the success of these assays. The use of primary cells derived from inducible knock-out mice, or the use of cells genetically altered by retroviral or lentiviral constructs, including fluorescence (gain-of-function) or by shRNA/siRNA knock-down or genome-editing CRISPR/Cas9-based methods (loss-of-function), has provided answers to specific hypotheses in angiogenesis processes. As opposed to in vitro assays, using in vivo assays, these hypotheses can be validated by immunohistochemical, histological and molecular biology procedures. Recently, the use of angiogenesis inhibitors in vivo has shown a potential effect in chronic liver disease, such as inhibiting the VEGF signaling pathway, combining treatments with VEGF and PDGF inhibitors directed against endothelial cells and pericytes, or gene therapy with cell-targeted molecule-targeted liposomal small interference RNAs (Table 1) [53][88][89]. Endogenous angiogenesis inhibitors using adenovirus-mediated gene transfer are also applied (Table 1) [56][90]. Gene expression inhibition studies employing in vivo loss-of-function models have shown that post-transcriptional mechanisms, regulated by cytoplasmic polyadenylation element binding proteins (CPEB), are essential for pathological angiogenesis in chronic liver disease, but dispensable for homeostasis of healthy vessels and physiological angiogenesis (Table 1) [30][54][55].
Despite the fact that in angiogenesis studies in vitro assays can be faster and reproducible, in vivo assays are physiologically more relevant. Moreover, in vitro assays do not represent the same conditions and do not allow study of the complex physiological interactions that occur in vivo. However, an in vivo angiogenic response also presents inconveniences, as it is a time-consuming and costly method, and it influences other parallel processes such as fibrogenesis and inflammation. Furthermore, angiogenesis image analysis requires advanced skills to avoid inter-observer variability. Finally, in vitro and in vivo models are essential to identify potential targets in neovascular processes in order to apply in new therapies in patients with chronic liver disease.
The Leading Models of Experimental ALD
ALD is one of the main causes of liver disease. The spectrum includes simple steatosis to cirrhosis, and it can lead to the development of end-stage HCC [91]. Unfortunately, there is no effective therapy. For the development of novel therapies, experimental animal models can provide more understanding of the mechanisms involved. The following is a short overview of the top three classic ALD experimental models and their hallmarks. First, several factors must be taken into account that could influence ALD models (Table 1) [31][92], such as gender, genetic background and age of mice. While female mice can develop alcoholic liver injury faster, they are less likely to progress to cirrhosis and HCC. For ALD models, the C57BL/6NCrl strain, due to its metabolism, is the most suitable. Moreover, the age recommended for onset of ALD models in mice is 8–11 weeks with a body weight above 19 g.
A-DW is a model for alcohol consumption in rodents, whereby the concentration of EtOH in the drinking water is gradually increased, and thereafter the animals are kept on the highest concentration throughout the study (up to 25% v/v, from eight up to 70 weeks). Other factors can be modified, such as opting to choose water or alcohol, or using multiple bottles with different alcohol concentrations or drinking in the dark. Despite the fact that this model is physiological, inexpensive, without significant mortality and with very simple animal husbandry, the strong natural aversion to alcohol of the animals results in reduced consumption and produces a reduced blood alcohol concentration (BAC) (50–70 mg/dL), thus inducing only moderate, clear steatosis and low elevations of ALT and AST without signs of fibrosis or inflammation [92][93].
The LDC diet is a liquid diet to which EtOH is added. This diet also contains necessary nutrients. At first, the EtOH is increased gradually from 1% to a concentration of 5.07% w/v (6.4% v/v) over a period of seven days. Next, mice are maintained with the highest EtOH concentration during a period which normally varies from 4 to 12 weeks. The control animal group is fed with LDC with the same isocaloric conditions as the LCD-EtOH diet. This diet has the advantage of being more time-efficient resulting in BAC ranging from 100 to 160 mg/dL. Preparation and management are straightforward, without specific equipment requirements and may be easily approved by local ethical committees. However, animals drink this diet when they are hungry and thirsty, and it is thus not completely physiological. The diet is freshly prepared every day, and the animals must be monitored during the entire process. As with AD-W, this diet also produces a mild elevation of serum transaminases and does not mimic advanced stages of human ALD such as cirrhosis and HCC (Table 1) [31][32].
There are different combinations of the LDC diet with secondary hepatic stressors or “second hits”, which have been widely used, producing models of progressive ALD. These combinations include the NIAAA model using 5% v/v LCD diet for ten days or eight weeks + single or multiple EtOH binges (5 g/kg) [33], the fibrotic model using moderate 2% LDC + CCl4 (1µL/g body weight, intraperitoneally (i.p.)), twice a week) (Table 1) [34], the HCC model with LCD (7–10 weeks) + DEN (40–100 mg/kg i.p.) [84], the LDC diet (8–10 weeks) + LPS (small-dose (1µg/g body weight) or high-dose (0.5 mg/kg, body weight)) [53] and the drug-induced liver injury model with the combination of LDC diet (4–6 weeks) + APAP (0.5–1 g/kg i.p.) [84].
In the intragastric ethanol infusion (IEI) method, mice are directly connected to an infusion pump with a catheter implanted into the stomach under aseptic conditions. Alcohol is added to the LDC diet and administered to the mice for a minimum period of six months. This method has the advantage of a sustained high BAC (250–500 mg/dL) and total control of nutritional intake. Furthermore, it can successfully produce advanced human ALD with the characteristic steatosis, apoptosis, central necrosis, inflammation, portal and bridging fibrosis. Nevertheless, this model bears the risk of infection and irritation, sometimes associated with dysbiosis. Therefore, it requires a high skill in implantation and considerable investment in equipment, with consequent difficulties in obtaining authorization from the local ethic committees [6][92].
In brief, none of the above-mentioned animal models can reproduce the features of human ALD due to the animals’ strong natural aversion to alcohol, high basal metabolic rate, fast catabolism of alcohol, spontaneous reduction in alcohol intake when acetaldehyde blood levels increase and absence of addictive behavior [94]. Nevertheless, these models can provide useful insights for novel therapeutic strategies.
Mouse Models for Non-Alcoholic Fatty Liver Disease
Non-alcoholic fatty liver disease (NAFLD) is the pathology induced in the liver characterized by fat deposition and hepatocyte steatosis. NASH is one of the stages in the spectrum of NAFLD that can progress to fibrosis, cirrhosis and, finally, to HCC. NASH is characterized by hepatocellular ballooning with fat vacuoles and the presence of inflammatory infiltrates. Unfortunately, as in other liver pathologies, not all animal models can replicate the full spectrum of human NASH. In order to study the mechanisms produced in NASH pathology and the potential therapeutic targets, the models most commonly used are as follows [42]: dietary models, such as HFD, high-fructose diet [43], cholesterol and cholate diet [37], MCD diet [95] and choline-deficient l-amino acid-defined (CDAA) diet [96]; genetic models, some of which act by promoting fat synthesis (leptin deficiency ob/ob mice [44], leptin receptor deficiency db/db mice or fa/fa in rat models [36][42]), while others act by inhibiting lipid peroxidation (peroxisome proliferator-activated receptor-α knock-out mice) [97] and impeding fat transport (ApoE knock-out mice) [35]; and chemical models, such as CCL4 [98], tetracycline [99] and streptozotocin in combination with HFD [100]. In brief, these models can reproduce different aspects of human NASH.
Mouse Models for Hepatocellular Carcinoma
HCC is the fifth leading cancer worldwide. Despite medical advances, treatment options are limited. HCC primarily occurs in the setting of chronic liver injury in a multistep process involving hepatitis (often associated with steatosis), liver fibrosis and cirrhosis. Animal models to mimic the different stages of human HCC are performed according to European/international animal welfare regulations as outlined elsewhere [101]. These models can be categorized into cell transplantation models, genetic models or chemical models alone or in combination with a second hit.
Cell transplantation HCC models consist of injection of hepatoma cells into recipient mice, either orthotopic in the liver (through intrahepatic or intrasplenic injection) or ectopic through subcutaneous application. Immune-deficient “nude-mice” are used for xenograft transplantation, enabling any kind of cell transplantation. Isograft transplantation consists of injection of hepatoma cells into recipient mice with identical genetic backgrounds. These models are fast and inexpensive, with easy management of the hepatoma cells before transplantation and without invasive monitoring of tumors. They are suitable for the testing of new drugs in HCC therapy. Nevertheless, these models do not allow study of all stages of progression of HCC, and in some models of tumors are occasionally rejected.
As a result of technological advances, genetic HCC mouse models, i.e., genetically engineered animal models of HCC, are increasingly developed [102]. Some of them enable study of steatosis, inflammation and fibrosis, such as liver-specific nuclear factor (NF)-κB essential modulator knock-out mice (NEMO) [103] or constitutive Mdr2-/- mice [104]. Patent regulations and material transfers are limitations of these models.
Of the genotoxic carcinogens, the DEN model of liver cancer is probably the best established chemical HCC model (Table 1) [45], whereby a single DEN injection in male mice at the age of exactly 14 days will give rise to small neoplastic lesions after 22 weeks and multinodular HCC after 40 weeks. The DEN injection model provides a high tumor incidence of 90%–100% and is easy to evaluate quantitatively by simply counting number and size of developed HCC nodules. As with other HCC models, the DEN chemical model cannot mimic the entire disease progression as in humans and testing of expensive new drugs is time-consuming. The DEN chemical model can also be complemented with a weekly injection of hepatotoxins, CCl4, as a second hit (Table 1) [46][47]. This model can mimic the human HCC processes, such as inflammation and fibrosis, after 30 days. However, it requires more effort, and measurement of tumors can be challenging due to changes in liver architecture.
The NASH/HCC model, a combination of NASH and weekly low-dose CCl4 injections (Table 1) [48], can reflect the progression of human fatty liver disease from simple steatosis to cancer within 24 weeks. With a high incidence within a short period of time, this model shows a close transcriptome similarity to human non-alcoholic fatty liver disease.
All these models provide important translational relevance, as they can mimic some of the human HCC mechanisms. For example, the c-Myc transgenic animal model is highly similar to human HCC with good prognosis, while DEN-derived tumors reflect human HCC in patients with poor survival (Table 1) [9][49]. Moreover, the NASH/HCC model develops similar mechanisms of human NAFLD) [48]. In brief, these models can be most valuable in the prognosis of chronic liver disease patients and thus decrease HCC incidence and mortality.
In Vivo Setup of Liver Disease Models for Surgery and Hemodynamic Assessments
Cirrhosis and portal hypertension cause significant morbidity and mortality [105]. Non-selective betablockers are currently the only medical therapy for portal hypertension, but have limited efficacy [106], and do not decrease liver fibrosis and intrahepatic vascular resistance. Thus, more effort is required to identify novel antifibrotic or anti-portal hypertensive drugs.
Importantly, the study of hepatic hemodynamics in animal models within the EU should follow the EU directive 2010/63/EU and national regulations [107][108]. Important recommendations for animal health monitoring, routine laboratory animal activities and animal welfare were specified by the Federation for Laboratory Animal Science Associations (FELASA) [109]. Furthermore, the principle of the 3Rs of Russell and Burch [110] must be considered in research with animals.
The appropriate handling of animal models of liver disease and the hemodynamic measurement requires experience and regular monitoring of the general health condition, such as of jaundice, ascites or hepatic encephalopathy during the induction period [39].
At evaluation of portal hypertension in vivo, the appropriate choice of anesthesia is another important aspect as it impacts on arterial blood pressure, heart rate (HR) and temperature. The equipment for anesthesia monitoring should include pulse oximeter, ECG and rectal temperature probe. The choice of anesthetics is based on pharmacodynamic characteristics, options for antagonization, administration routes and differences in sensitivity related to sex, animal species and strain [111]. In addition, the planned interventions, such as vascular cannulations must be taken into consideration, which can be surgically challenging and require specific hemodynamic equipment [51]. A typical hemodynamic assessment of the portal hypertensive syndrome requires about 45 min per animal for the simultaneous acquisition of several important systemic, splanchnic and portal hemodynamic parameters.
A single injection of anesthesia requires less equipment, is relatively cheap, and does not require extensive skill with animal handling. However, the hemodynamic assays described below are very sensitive to the depth of anesthesia, which is challenging by a single injection and moreover could interfere with the results of the measurements. In contrast, the combination of inhalation anesthesia supported by intubation and injective anesthetics allows for an improved control and safety of the procedures.
For a detailed evaluation of the various hemodynamic parameters characterizing the portal hypertension syndrome and some other surgical interventions, using mice may represent a limitation due to their small body size that requires a high level of expertise and degree of technical expertise. This is probably the most important reason why usually rats are used when several hemodynamic parameters must be simultaneously obtained.
The main hemodynamic parameters to characterize the portal hypertension syndrome are portal pressure (PP), mean arterial pressure (MAP), HR, superior mesenteric artery blood flow (SMABF) and portal vein blood flow (PVBF). The ratio between MAP and HR represents a hyperdynamic index, which reflects the severity of hyperdynamic circulation caused by peripheral/splanchnic vasodilation. MAP/HR are usually invasively measured via cannulation of the femoral or carotid artery following surgical dissection and using an intravascular catheter connected to a pressure transducer. After median laparotomy, SMABF can be measured by perivascular, non-constrictive ultrasound flow probes at the superior mesenteric artery as a surrogate parameter for splanchnic vasodilation. A similar, but different-sized flow probe is available for measurement of PVBF at the portal vein. Importantly, flow probes must be calibrated for the viscosity of blood, and their size has to be chosen according to the targeted vessel size. Portal pressure as main readout parameter is assessed by a direct cannulation of a suitable mesenteric venous blood vessel, usually at the junction of the ileocolic vein by an intravascular catheter. Portal pressure is recorded via a pressure transducer after careful advancement of the catheter to the portal vein close to the liver hilum. Intrahepatic vascular resistance can be calculated by PP and PVBF.
In patients, portal pressure can also be measured invasively via the puncture of the portal vein; however, nowadays, mostly the indirect measurement via the hepatic venous pressure gradient (HVPG) assessed via transjugular liver vein catheterization is used. Invasive assessment of arterial pressure and HR is also possible via an arterial line as in an intensive care unit. In addition, splanchnic and portal blood flow may be semi-quantitatively assessed by percutaneous Doppler ultrasound. Novel dynamic contrast-enhanced CT- or MRI-based cross-sectional imaging methods are currently being developed that may allow for a quantification of blood flow in various splanchnic and portal-venous and arterial blood vessels.
Conclusions
Advanced technologies in molecular and cellular research have rapidly evolved to meet the demands of clinical applications involving diagnostics and therapeutics in liver diseases.
Extensive research is required to select the appropriate model for specific liver research issues. Any experiment or model for research purposes must be meticulously planned and should combine different in vivo and in vitro models (Figure 2). While the currently existing knowledge gaps in the study of liver pathophysiology can be addressed with all these methodologies, future techniques and methods for different fields must, nevertheless, be continuously adapted for liver research (Supplementary Table S1).
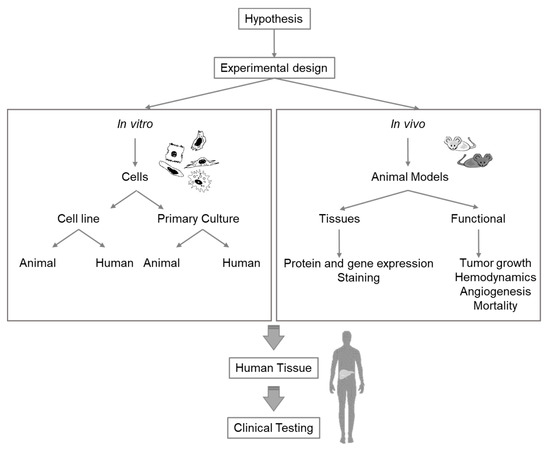
Figure 2. Diagram of considerations for selection of the appropriate experimental model for liver research.
Supplementary Materials: Supplementary materials can be found at https://www.mdpi.com/1422-0067/21/6/2027/s1.
Funding: The authors were supported by grants from the Deutsche Forschungsgemeinschaft (SFB TRR57, CRC1382). The MICROB-PREDICT, GALAXY and LIVERHOPE projects have received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreements numbers 825694, 668031 and 731875 respectively. The manuscript reflects only the authors’ views, and the European Commission is not responsible for any use that may be made of the information it contains. The funders had no influence on study design, data collection and analysis, decision to publish or preparation of the manuscript. The conference was supported by grants from the Deutsche Forschungsgemeinschaft (SFB TRR57) (20.000€), EASL (10.000€) and EFCLIF (10.000€). An unrestricted grant of 850€ was obtained by Bayer.
Conflicts of Interest: The authors have no conflicts of interest.
Abbreviations
| A-DW | Alcohol in drinking water |
| ALD | Alcoholic liver disease |
| APAP | Acetaminophen |
| BAC | Blood alcohol concentration |
| BEC | Biliary epithelial cells |
| CCL4 | Carbon tetrachloride |
| CDAA | Choline-deficient l-amino acid-defined |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| DEN | N-nitrosodiethylamine |
| FACS | Fluorescence-activated cell sorting |
| FELASA | Federation for Laboratory Animal Science Associations |
| GGT | Γ-Glutamyl-transpeptidase |
| HCC | Hepatocellular carcinoma |
| HFD | High-fat diet |
| HR | Heart rate |
| HSCs | Hepatic stellate cells |
| HVPG | Hepatic venous pressure gradient |
| I.m. | Intramuscularly |
| I.p. | Intraperitoneally |
| IBDU | Intrahepatic bile duct units |
| IEI | Intragastric ethanol infusion |
| iPSCs | Induced pluripotent stem cells |
| KCs | Kupffer cells |
| LDC | Lieber–DeCarli |
| LPS | Lipopolysaccharide |
| MACS | Magnetic activated cell sorting |
| MAP | Mean arterial pressure |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NEMO | Nuclear factor (NF)-κB essential modulator |
| NIAAA | Chronic-plus-binge alcohol feeding model |
| PAPPA | Pregnancy-associated plasma protein A |
| PP | Portal pressure |
| PVBF | Portal vein blood flow |
| SMABF | Superior mesenteric artery blood flow |
| WD | Western diet |
This entry is adapted from the peer-reviewed paper 10.3390/ijms21062027
References
- Kaori Endo-Umeda; Makoto Makishima; Liver X Receptors Regulate Cholesterol Metabolism and Immunity in Hepatic Nonparenchymal Cells.. International Journal of Molecular Sciences 2019, 20, 5045, 10.3390/ijms20205045.
- G Pott; J Rauterberg; B Voss; U Gerlach; Connective tissue components of the normal and fibrotic human liver. II. Clinical aspects.. 1982, 60, , .
- Kyo-Sang Yoo; Woo Taek Lim; Ho Soon Choi; Biology of Cholangiocytes: From Bench to Bedside. Gut and Liver 2016, 10, 687-698, 10.5009/gnl16033.
- Oliver Krenkel; Jana Hundertmark; Ali T Abdallah; Marlene Kohlhepp; Tobias Puengel; Tilmann Roth; Diogo Philippini Pontual Branco; Jana C Mossanen; Tom Luedde; Christian Trautwein; et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut 2019, 69, 551-563, 10.1136/gutjnl-2019-318382.
- Robert Huebert; Sampaziotis F; Justin Aw; Tysoe Oc; Sawiak S; Godfrey Em; Upponi Ss; Gieseck Rl; De Brito Mc; Berntsen Nl; et al. Faculty of 1000 evaluation for Reconstruction of the mouse extrahepatic biliary tree using primary human extrahepatic cholangiocyte organoids.. F1000 - Post-publication peer review of the biomedical literature 2017, 23, , 10.3410/f.727772304.793536483.
- H. Tsukamoto; R. D. Reidelberger; S. W. French; C. Largman; Long-term cannulation model for blood sampling and intragastric infusion in the rat. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 1984, 247, R595-R599, 10.1152/ajpregu.1984.247.3.r595.
- Maximilian J Brol; Felicitas Rösch; Robert Schierwagen; Fernando Magdaleno; Frank Erhard Uschner; Steffen Manekeller; Alexander Queck; Katharina Schwarzkopf; Margarete Odenthal; Uta Drebber; et al. Combination of CCl4 with alcoholic and metabolic injuries mimics human liver fibrosis.. American Journal of Physiology-Gastrointestinal and Liver Physiology 2019, 317, G182-G194, 10.1152/ajpgi.00361.2018.
- Matthias Bartneck; Peter L. Schrammen; Diana Möckel; Olivier Govaere; Anke Liepelt; Oliver Krenkel; Can Ergen; Misti Vanette McCain; Dirk Eulberg; Tom Luedde; et al. The CCR2+ Macrophage Subset Promotes Pathogenic Angiogenesis for Tumor Vascularization in Fibrotic Livers. Cellular and Molecular Gastroenterology and Hepatology 2019, 7, 371-390, 10.1016/j.jcmgh.2018.10.007.
- Corrigendum to . , , , .
- Albert Geerts; Toshiro Niki; Karine Hellemans; Dirk De Craemer; Kit Van Den Berg; Jean-Marc Lazou; Geert Stange; Marnix Van De Winkel; Pieter De Bleser; Purification of rat hepatic stellate cells by side scatter-activated cell sorting. Hepatology 1998, 27, 590-598, 10.1002/hep.510270238.
- Sabine Weiskirchen; Carmen G. Tag; Sibille Sauer-Lehnen; Frank Tacke; Ralf Weiskirchen; Isolation and Culture of Primary Murine Hepatic Stellate Cells. Advanced Structural Safety Studies 2017, 1627, 165-191, 10.1007/978-1-4939-7113-8_11.
- Frank Tacke; Ralf Weiskirchen; Update on hepatic stellate cells: pathogenic role in liver fibrosis and novel isolation techniques. Expert Review of Gastroenterology & Hepatology 2012, 6, 67-80, 10.1586/egh.11.92.
- Jana C. Mossanen; Oliver Krenkel; Can Ergen; Olivier Govaere; Anke Liepelt; Tobias Puengel; Felix Heymann; Sandra Kalthoff; Eric Lefebvre; Dirk Eulberg; et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 2016, 64, 1667-1682, 10.1002/hep.28682.
- A. Mennone; Domenico Alvaro; W. Cho; J. L. Boyer; Isolation of small polarized bile duct units.. Proceedings of the National Academy of Sciences 1995, 92, 6527-6531, 10.1073/pnas.92.14.6527.
- Li Yang; Ronald A. Faris; Uglas C. Hixson; Characterization of a mature bile duct antigen expressed on a subpopulation of biliary ductular cells but absent from oval cells. Hepatology 1993, 18, 357-366, 10.1002/hep.1840180220.
- Mühlbauer, M.; Weiss, T.S.; Thasler, W.E.; Gelbmann, C.M.; Schnabl, B.; Schölmerich, J.; Hellerbrand, C. LPS-mediated NFkappaB activation varies between activated human hepatic stellate cells from different donors. Biochem. Biophys. Res. Commun. 2004, 325, 191–197. [Google Scholar] [CrossRef]
- Claus Hellerbrand; Hepatic stellate cells—the pericytes in the liver. Pflügers Archiv - European Journal of Physiology 2013, 465, 775-778, 10.1007/s00424-012-1209-5.
- Tatjana Seitz; Kim Freese; Peter Dietrich; Wolfgang Erwin Thasler; Anja Bosserhoff; Claus Hellerbrand; Fibroblast Growth Factor 9 is expressed by activated hepatic stellate cells and promotes progression of hepatocellular carcinoma. Scientific Reports 2020, 10, 1-9, 10.1038/s41598-020-61510-4.
- Theresa Meyer; Andreas Koch; Eva-Vanessa Ebert; Barbara Czech; Martina Mueller; Anja Bosserhoff; Sven Arke Lang; Claus Hellerbrand; Effect of melanoma cells on proliferation and migration of activated hepatic stellate cells in vitro. Pathology - Research and Practice 2017, 213, 400-404, 10.1016/j.prp.2016.12.014.
- Hella Wobser; Christoph Dorn; Thomas S. Weiss; Thomas Amann; Cornelius Bollheimer; Roland Büttner; Jürgen Schölmerich; Claus Hellerbrand; Roland B; [Uuml]; et al. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell Research 2009, 19, 996-1005, 10.1038/cr.2009.73.
- Oliver Krenkel; Tobias Puengel; Olivier Govaere; Ali T. Abdallah; Jana C. Mossanen; Marlene Kohlhepp; Anke Liepelt; Eric Lefebvre; Tom Luedde; Claus Hellerbrand; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis.. Hepatology 2018, 67, 1270-1283, 10.1002/hep.29544.
- Charlotte L. Scott; Wouter T’Jonck; Liesbet Martens; Helena Todorov; Dorine Sichien; Bieke Soen; Johnny Bonnardel; Sofie De Prijck; Niels Vandamme; Robrecht Cannoodt; et al. The Transcription Factor ZEB2 Is Required to Maintain the Tissue-Specific Identities of Macrophages.. Immunity 2018, 49, 312-325.e5, 10.1016/j.immuni.2018.07.004.
- Sonya A. MacParland; Jeff C. Liu; Xue-Zhong Ma; Brendan Innes; Agata M. Bartczak; Blair K. Gage; Justin Manuel; Nicholas Khuu; Juan Echeverri; Ivan Linares; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nature Communications 2018, 9, 4383, 10.1038/s41467-018-06318-7.
- Neta Lavon; Ofra Yanuka; Nissim Benvenisty; Differentiation and isolation of hepatic-like cells from human embryonic stem cells. Differentiation 2004, 72, 230-238, 10.1111/j.1432-0436.2004.07205002.x.
- Robert E. Schwartz; Jonathan L. Linehan; Matthew S. Painschab; Wei-Shou Hu; Catherine M. Verfaillie; Dan S. Kaufman; Defined Conditions for Development of Functional Hepatic Cells from Human Embryonic Stem Cells. Stem Cells and Development 2005, 14, 643-655, 10.1089/scd.2005.14.643.
- Arefeh Ghodsizadeh; Adeleh Taei; Mehdi Totonchi; Ali Seifinejad; Hamid Gourabi; Behshad Pournasr; Nasser Aghdami; Reza Malekzadeh; Navid Almadani; Ghasem Hosseini Salekdeh; et al. Generation of Liver Disease-Specific Induced Pluripotent Stem Cells Along with Efficient Differentiation to Functional Hepatocyte-Like Cells. Stem Cell Reviews and Reports 2010, 6, 622-632, 10.1007/s12015-010-9189-3.
- Romina Fiorotto; Mariangela Amenduni; Valeria Mariotti; Luca Fabris; Carlo Spirli; Mario Strazzabosco; Liver diseases in the dish: iPSC and organoids as a new approach to modeling liver diseases.. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2018, 1865, 920-928, 10.1016/j.bbadis.2018.08.038.
- Patricio Godoy; Nicola J. Hewitt; Ute Albrecht; Melvin E. Andersen; Nariman Ansari; Sudin Bhattacharya; Johannes Georg Bode; Jennifer Bolleyn; Christoph Borner; Jan Böttger; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Archives of Toxicology 2013, 87, 1315-530, 10.1007/s00204-013-1078-5.
- Marta Ramirez; Angiogenesis in Liver Disease Research Group; Mercedes Fernandez; 08036 Barcelona Rossello 149-153; Pathological Angiogenesis: The New Culprit behind Chronic Liver Disease. OBM Hepatology and Gastroenterology 2019, 3, 1-1, 10.21926/obm.hg.1903030.
- Ester García-Pras; Javier Gallego; Laura Coch; Marc Mejias; Gonzalo Fernandez-Miranda; Ricardo Pardal; Jaime Bosch; Raúl Méndez; Mercedes Fernandez; Role and therapeutic potential of vascular stem/progenitor cells in pathological neovascularisation during chronic portal hypertension. Gut 2016, 66, 1306-1320, 10.1136/gutjnl-2015-311157.
- Feifei Guo; Kang Zheng; Raquel Benedé-Ubieto; Francisco Javier Cubero; Yulia A. Nevzorova; The Lieber-DeCarli Diet-A Flagship Model for Experimental Alcoholic Liver Disease. Alcoholism: Clinical and Experimental Research 2018, 42, 1828-1840, 10.1111/acer.13840.
- Liquid Diet Technique of ethanol administration: 1989 update. Alcohol Alcohol. 1989, 24, 197–211.
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Sameek Roychowdhury; Dian J. Chiang; Megan R. McMullen; Laura E. Nagy; Moderate, chronic ethanol feeding exacerbates carbon tetrachloride-induced hepatic fibrosis via hepatocyte-specific hypoxia-inducible factor 1α. Pharmacology Research & Perspectives 2014, 2, e00061, 10.1002/prp2.61.
- Robert Schierwagen; Lara Maybüchen; Sebastian Zimmer; Kanishka Hittatiya; Christer Bäck; Sabine Klein; Frank E. Uschner; Winfried Reul; Peter Boor; Georg Nickenig; et al. Seven weeks of Western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Scientific Reports 2015, 5, 12931, 10.1038/srep12931.
- Priya Handa; Vicki Morgan-Stevenson; Bryan D. Maliken; James E. Nelson; Shenna Washington; Mark Westerman; Matthew M. Yeh; Kris V Kowdley; Iron overload results in hepatic oxidative stress, immune cell activation, and hepatocellular ballooning injury, leading to nonalcoholic steatohepatitis in genetically obese mice. American Journal of Physiology-Gastrointestinal and Liver Physiology 2016, 310, G117-G127, 10.1152/ajpgi.00246.2015.
- Montserrat Marí; Francisco Caballero; Anna Colell; Albert Morales; Juan Caballería; Ana Fernandez; Carlos Enrich; José C. Fernándezcheca; Carmen García-Ruiz; Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metabolism 2006, 4, 185-198, 10.1016/j.cmet.2006.07.006.
- Frank Tacke; Targeting hepatic macrophages to treat liver diseases. Journal of Hepatology 2017, 66, 1300-1312, 10.1016/j.jhep.2017.02.026.
- Howard Spiro; Animal models of portal hypertension. Gastroenterology 1989, 96, 1223-1224, 10.1016/0016-5085(89)91656-9.
- Christer Baeck; Alexander Wehr; Karlin Raja Karlmark; Felix Heymann; Mihael Vucur; Nikolaus Gassler; Sebastian Huss; Sven Klussmann; Dirk Eulberg; Tom Luedde; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2011, 61, 416-426, 10.1136/gutjnl-2011-300304.
- Claudia Von Montfort; Nuria Matias; Ana Fernandez; Raquel Fucho; Laura Conde De La Rosa; María Luz Martínez-Chantar; José M Mato; Keigo Machida; Hidekazu Tsukamoto; Michael P. Murphy; et al. Mitochondrial GSH determines the toxic or therapeutic potential of superoxide scavenging in steatohepatitis.. Journal of Hepatology 2012, 57, 852-9, 10.1016/j.jhep.2012.05.024.
- Mikhaïl A. Van Herck; Luisa Vonghia; S. Francque; Animal Models of Nonalcoholic Fatty Liver Disease—A Starter’s Guide. Nutrients 2017, 9, 1072, 10.3390/nu9101072.
- Longato, L. Non-alcoholic fatty liver disease (NAFLD): A tale of fat and sugar? Fibrogenes. Tissue Repair 2013, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Per Lindström; The Physiology of Obese-Hyperglycemic Mice [ob/ob Mice]. The Scientific World Journal 2007, 7, 666-685, 10.1100/tsw.2007.117.
- Christian Liedtke; Tom Luedde; Tilman Sauerbruch; D. Scholten; K.L. Streetz; Frank Tacke; René H Tolba; C. Trautwein; Jonel Trebicka; Ralf Weiskirchen; et al. Experimental liver fibrosis research: update on animal models, legal issues and translational aspects. Fibrogenesis & Tissue Repair 2013, 6, 19-19, 10.1186/1755-1536-6-19.
- Tom Luedde; Naiara Beraza; Vasileios Kotsikoris; Geert Van Loo; Arianna Nenci; Rita De Vos; Tania Roskams; Christian Trautwein; Manolis Pasparakis; Deletion of NEMO/IKKγ in Liver Parenchymal Cells Causes Steatohepatitis and Hepatocellular Carcinoma. Cancer Cell 2007, 11, 119-132, 10.1016/j.ccr.2006.12.016.
- J.J.M. Smit; A.H. Schinkel; R.P.J.Oude Elferink; A.K. Groen; E. Wagenaar; L. Van Deemter; C.A.A.M. Mol; R. Ottenhoff; N.M.T. Van Der Lugt; M.A. Van Roon; et al. Homozygous disruption of the murine MDR2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 1993, 75, 451-462, 10.1016/0092-8674(93)90380-9.
- Tolba, R.; Kraus, T.; Liedtke, C.; Schwarz, M.; Weiskirchen, R. Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab. Anim. 2015, 49, 59–69. [Google Scholar] [CrossRef]
- Uehara, T.; Pogribny, I.P.; Rusyn, I. The DEN and CCl4 -Induced Mouse Model of Fibrosis and Inflammation-Associated Hepatocellular Carcinoma. Curr. Protoc. Pharmacol. 2014, 66, 1–10. [Google Scholar] [CrossRef]
- S Klein; C Hinüber; K Hittatiya; R Schierwagen; Fe Uschner; Cp Strassburg; Hp Fischer; U Spengler; J Trebicka; Novel rat model of repetitive portal venous embolization mimicking human non-cirrhotic idiopathic portal hypertension. Zeitschrift für Gastroenterologie 2016, 54, 1343-1404, 10.1055/s-0036-1597369.
- Philipp Königshofer; Ksenia Brusilovskaya; Philipp Schwabl; Bruno Podesser; Michael Trauner; Thomas Reiberger; Invasive Hemodynamic Characterization of the Portal-hypertensive Syndrome in Cirrhotic Rats. Journal of Visualized Experiments 2018, , , 10.3791/57261.
- Sabine Klein; Johanna Rick; Jennifer Lehmann; Robert Schierwagen; Irela Gretchen Schierwagen; Len Verbeke; Kanishka Hittatiya; Frank Erhard Uschner; Steffen Manekeller; Christian P Strassburg; et al. Janus-kinase-2 relates directly to portal hypertension and to complications in rodent and human cirrhosis. Gut 2015, 66, 145-155, 10.1136/gutjnl-2015-309600.
- Marc Mejias; Carolina Tiani; Rosa Miquel; Mercedes Fernandez; Ester García-Pras; Jaime Bosch; Beneficial effects of sorafenib on splanchnic, intrahepatic, and portocollateral circulations in portal hypertensive and cirrhotic rats†. Hepatology 2008, 49, 1245-1256, 10.1002/hep.22758.
- Vittorio Calderone; Javier Gallego; Gonzalo Fernandez-Miranda; Ester García-Pras; Carlos Maillo; Annalisa Berzigotti; Marc Mejias; Felice-Alessio Bava; Ana Angulo-Urarte; Mariona Graupera; et al. Sequential Functions of CPEB1 and CPEB4 Regulate Pathologic Expression of Vascular Endothelial Growth Factor and Angiogenesis in Chronic Liver Disease. Gastroenterology 2016, 150, 982-997.e30, 10.1053/j.gastro.2015.11.038.
- Carlos Maillo; Judit Martín; David Sebastián; María Isabel Hernández-Alvarez; Mar García-Rocha; Oscar Reina; Antonio Zorzano; Mercedes Fernandez; Raúl Méndez; Circadian- and UPR-dependent control of CPEB4 mediates a translational response to counteract hepatic steatosis under ER stress. Nature 2017, 19, 94-105, 10.1038/ncb3461.
- Laura Coch; Marc Mejias; Annalisa Berzigotti; Javier Gallego; Jaime Bosch; Raúl Méndez; Mercedes Fernandez; Ester García-Pras; Disruption of negative feedback loop between vasohibin-1 and vascular endothelial growth factor decreases portal pressure, angiogenesis, and fibrosis in cirrhotic rats. Hepatology 2014, 60, 633-647, 10.1002/hep.26995.
- A Blouin; R P Bolender; E R Weibel; Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. Journal of Cell Biology 1977, 72, 441-455, .
- Ryan J. Schulze; Micah B. Schott; Carol A. Casey; Pamela L. Tuma; Mark A. McNiven; The cell biology of the hepatocyte: A membrane trafficking machine. Journal of Cell Biology 2019, 218, 2096-2112, 10.1083/jcb.201903090.
- A. L. Dounce; THE PREPARATION OF ISOLATED CELL NUCLEI OF RAT LIVER. Science 1944, 99, 512-513, 10.1126/science.99.2582.512.
- Victoria Kegel; Daniela Deharde; Elisa Pfeiffer; Katrin Zeilinger; Daniel Seehofer; Georg Damm; Protocol for Isolation of Primary Human Hepatocytes and Corresponding Major Populations of Non-parenchymal Liver Cells. Journal of Visualized Experiments 2016, , e53069-e53069, 10.3791/53069.
- M. Teresa Donato; Miguel Bolonio; Estefanía Cabezas; María Pelechá; Eugenia Pareja; Anna Domènech; José V. Castell; M. José Gómez-Lechón; Laia Tolosa; Improved in vivo efficacy of clinical-grade cryopreserved human hepatocytes in mice with acute liver failure. Cytotherapy 2020, 22, 114-121, 10.1016/j.jcyt.2019.12.005.
- Behshad Pournasr; Stephen Duncan; Modeling Inborn Errors of Hepatic Metabolism Using Induced Pluripotent Stem Cells.. Arteriosclerosis, Thrombosis, and Vascular Biology 2017, 37, 1994-1999, 10.1161/ATVBAHA.117.309199.
- James L. Corbett; Stephen Duncan; iPSC-Derived Hepatocytes as a Platform for Disease Modeling and Drug Discovery.. Frontiers in Medicine 2019, 6, 265, 10.3389/fmed.2019.00265.
- Andrés J M Ferreri; Gerald Illerhaus; Emanuele Zucca; Franco Cavalli; International Extranodal Lymphoma Study group; on behalf of the International Extranodal Lymphoma Study Group; Flows and flaws in primary central nervous system lymphoma.. Nature Reviews Clinical Oncology 2010, 7, 1-2, 10.1038/nrclinonc.2010.9-c1.
- Hans-Theo Schon; Matthias Bartneck; Erawan Borkham-Kamphorst; Jacob Nattermann; Twan Lammers; Frank Tacke; Ralf Weiskirchen; Pharmacological Intervention in Hepatic Stellate Cell Activation and Hepatic Fibrosis. Frontiers in Pharmacology 2016, 7, 536, 10.3389/fphar.2016.00033.
- H.F.J. Hendriks; W.A.M.M. Verhoofstad; A. Brouwer; A.M. De Leeuw; D.L. Knook; Perisinusoidal fat-storing cells are the main vitamin A storage sites in rat liver. Experimental Cell Research 1985, 160, 138-149, 10.1016/0014-4827(85)90243-5.
- Samuele De Minicis; Ekihiro Seki; Hiroshi Uchinami; Johannes Kluwe; Yonghui Zhang; David A. Brenner; Robert F. Schwabe; Gene Expression Profiles During Hepatic Stellate Cell Activation in Culture and In Vivo. Gastroenterology 2007, 132, 1937-1946, 10.1053/j.gastro.2007.02.033.
- Ryan E. Feaver; Banumathi K. Cole; Mark J. Lawson; Stephen A. Hoang; Svetlana Marukian; Brett R. Blackman; Robert A. Figler; Arun J. Sanyal; Brian R. Wamhoff; Ajit Dash; et al. Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis.. JCI Insight 2016, 1, e90954, 10.1172/jci.insight.90954.
- Marcus Mühlbauer; Anja Bosserhoff; Arndt Hartmann; Wolfgang E Thasler; Thomas S. Weiss; Hans Herfarth; Guntram Lock; Jürgen Schölmerich; Claus Hellerbrand; A novel MCP-1 gene polymorphism is associated with hepatic MCP-1 expression and severity of HCV-related liver disease.. Gastroenterology 2003, 125, 1085-1093, 10.1016/s0016-5085(03)01213-7.
- Julia C. Engelmann; Thomas Amann; Birgitta Ott-Rötzer; Margit Nützel; Yvonne Reinders; Jörg Reinders; Wolfgang E. Thasler; Theresa Kristl; Andreas Teufel; Christian G Huber; et al. Causal Modeling of Cancer-Stromal Communication Identifies PAPPA as a Novel Stroma-Secreted Factor Activating NFκB Signaling in Hepatocellular Carcinoma. PLOS Computational Biology 2015, 11, e1004293, 10.1371/journal.pcbi.1004293.
- Oliver Krenkel; Frank Tacke; Liver macrophages in tissue homeostasis and disease. Nature Reviews Immunology 2017, 17, 306-321, 10.1038/nri.2017.11.
- Prakash Ramachandran; R. Dobie; J. R. Wilson-Kanamori; E. F. Dora; B. E. P. Henderson; N. T. Luu; J. R. Portman; K. P. Matchett; M. Brice; J. A. Marwick; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512-518, 10.1038/s41586-019-1631-3.
- Matthias Bartneck; Klaudia Theresa Warzecha; Carmen Gabriele Tag; Sibille Sauer-Lehnen; Felix Heymann; Christian Trautwein; Ralf Weiskirchen; Frank Tacke; Isolation and Time Lapse Microscopy of Highly Pure Hepatic Stellate Cells. Analytical Cellular Pathology 2015, 2015, 1-13, 10.1155/2015/417023.
- Johnny Bonnardel; Wouter T’Jonck; Djoere Gaublomme; Robin Browaeys; Charlotte L. Scott; Liesbet Martens; Bavo Vanneste; Sofie De Prijck; Sergei A. Nedospasov; Anna Kremer; et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche.. Immunity 2019, 51, 638-654.e9, 10.1016/j.immuni.2019.08.017.
- Maureen E. Taylor; Tom Snelling; David F Smith; Kurt Drickamer; Absence of a human ortholog of rodent Kupffer cell galactose-binding receptor encoded by the CLEC4f gene.. Glycobiology 2019, 29, 332-345, 10.1093/glycob/cwy113.
- Mario Strazzabosco; Luca Fabris; Functional anatomy of normal bile ducts.. The Anatomical Record 2008, 291, 653-60, 10.1002/ar.20664.
- Luca Fabris; Carlo Spirli; Massimiliano Cadamuro; Romina Fiorotto; Mario Strazzabosco; Emerging concepts in biliary repair and fibrosis.. American Journal of Physiology-Gastrointestinal and Liver Physiology 2017, 313, G102-G116, 10.1152/ajpgi.00452.2016.
- Carlo Spirli; Luca Fabris; Elena Duner; Romina Fiorotto; Giorgio Ballardini; Tania Roskams; Nicholas F. LaRusso; Aurelio Sonzogni; Lajos Okolicsanyi; Mario Strazzabosco; et al. Cytokine-stimulated nitric oxide production inhibits adenylyl cyclase and cAMP-dependent secretion in cholangiocytes. Gastroenterology 2003, 124, 737-753, 10.1053/gast.2003.50100.
- Carlo Spirlì; Romina Fiorotto; Lei Song; Joseph Santos-Sacchi; Lajos Okolicsanyi; Sara Masier; Loretta Rocchi; Mariapia Vairetti; Marina De Bernard; Saida Melero; et al. Glibenclamide stimulates fluid secretion in rodent cholangiocytes through a cystic fibrosis transmembrane conductance regulator-independent mechanism.. Gastroenterology 2005, 129, 220-233, 10.1053/j.gastro.2005.03.048.
- Carlo Spirlı̀; Michael H. Nathanson; Romina Fiorotto; Elena Duner; Lee A. Denson; Juana M Sanz; Francesco Di Virgilio; Lajos Okolicsanyi; Fabio Casagrande; Mario Strazzabosco; et al. Proinflammatory Cytokines Inhibit Secretion in Rat Bile Duct Epithelium. Gastroenterology 2001, 121, 156-169, 10.1053/gast.2001.25516.
- Luca Fabris; Massimiliano Cadamuro; Romina Fiorotto; Tania Roskams; Carlo Spirli; Saida Melero; Aurelio Sonzogni; Ruth Joplin; Lajos Okolicsanyi; Mario Strazzabosco; et al. Effects of angiogenic factor overexpression by human and rodent cholangiocytes in polycystic liver diseases. Hepatology 2006, 43, 1001-1012, 10.1002/hep.21143.
- Romina Fiorotto; Ambra Villani; Antonis Kourtidis; Roberto Scirpo; Mariangela Amenduni; Peter J. Geibel; Massimiliano Cadamuro; Carlo Spirli; Panos Z. Anastasiadis; Mario Strazzabosco; et al. The cystic fibrosis transmembrane conductance regulator controls biliary epithelial inflammation and permeability by regulating Src tyrosine kinase activity.. Hepatology 2016, 64, 2118-2134, 10.1002/hep.28817.
- Ruth Joplin; A. J. Strain; J. M. Neuberger; Biliary epithelial cells from the liver of patients with primary biliary cirrhosis: Isolation, characterization, and short-term culture. The Journal of Pathology 1990, 162, 255-260, 10.1002/path.1711620312.
- A. J. Strain; L. Wallace; R. Joplin; Y. Daikuhara; T. Ishii; D. A. Kelly; J. M. Neuberger; Characterization of biliary epithelial cells isolated from needle biopsies of human liver in the presence of hepatocyte growth factor.. The American Journal of Pathology 1995, 146, 537-545, .
- Romina Fiorotto; Mariangela Amenduni; Valeria Mariotti; Luca Fabris; Carlo Spirli; Mario Strazzabosco; Src kinase inhibition reduces inflammatory and cytoskeletal changes in ΔF508 human cholangiocytes and improves cystic fibrosis transmembrane conductance regulator correctors efficacy.. Hepatology 2018, 67, 972-988, 10.1002/hep.29400.
- Mercedes Fernandez; Molecular pathophysiology of portal hypertension. Hepatology 2015, 61, 1406-1415, 10.1002/hep.27343.
- Patrycja Nowak-Sliwinska; Kari Alitalo6; Elizabeth Allen; Andrey Anisimov; Alfred C. Aplin; Robert Auerbach; Hellmut G. Augustin; David O. Bates; Judy R. Van Beijnum; R. Hugh F. Bender; et al. Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis 2018, 21, 425-532, 10.1007/s10456-018-9613-x.
- Thomas Reiberger; Bernhard Angermayr; Philipp Schwabl; Natascha Rohr-Udilova; Markus Mitterhauser; Alfred Gangl; Markus Peck-Radosavljevic; Sorafenib attenuates the portal hypertensive syndrome in partial portal vein ligated rats. Journal of Hepatology 2009, 51, 865-873, 10.1016/j.jhep.2009.06.024.
- Javier Gallego; Ester Garcia-Pras; Marc Mejias; Nuria Pell; Ute Schaeper; Mercedes Fernandez; Therapeutic siRNA targeting endothelial KDR decreases portosystemic collateralization in portal hypertension.. Scientific Reports 2017, 7, 14791, 10.1038/s41598-017-14818-7.
- Marc Mejias; L. Coch; A. Berzigotti; Ester García-Pras; J. Gallego; J. Bosch; M. Fernández; Antiangiogenic and antifibrogenic activity of pigment epithelium-derived factor (PEDF) in bile duct-ligated portal hypertensive rats. Gut 2014, 64, 657-666, 10.1136/gutjnl-2014-307138.
- Amir Anushiravani; Sadaf Ghajarieh Sepanlou; Burden of Liver Diseases: A Review from Iran. Middle East Journal of Digestive Diseases 2019, 11, 189-191, 10.15171/mejdd.2019.147.
- Nympha B. D'souza El-Guindy; Elizabeth J. Kovacs; Philippe De Witte; Claudia Spies; John M. Littleton; Willem J. S. De Villiers; Amanda J. Lott; Timothy P. Plackett; Nadine Lanzke; Gary G. Meadows; et al. Laboratory models available to study alcohol-induced organ damage and immune variations: choosing the appropriate model.. Alcoholism: Clinical and Experimental Research 2010, 34, 1489-511, 10.1111/j.1530-0277.2010.01234.x.
- Elizabeth Brandon-Warner; Laura W. Schrum; C. Max Schmidt; Iain H McKillop; Rodent models of alcoholic liver disease: of mice and men.. Alcohol 2012, 46, 715-25, 10.1016/j.alcohol.2012.08.004.
- Benedicte Delire; Peter Starkel; Isabelle Leclercq; Animal Models for Fibrotic Liver Diseases: What We Have, What We Need, and What Is under Development. Journal of Clinical and Translational Hepatology 2015, 3, 53-66, 10.14218/JCTH.2014.00035.
- Hiroko Itagaki; Kazuhiko Shimizu; Shunichi Morikawa; Kenji Ogawa; Taichi Ezaki; Morphological and functional characterization of non-alcoholic fatty liver disease induced by a methionine-choline-deficient diet in C57BL/6 mice. International journal of clinical and experimental pathology 2013, 6, 2683-2696, .
- Kirstine S. Tølbøl; Birgit Stierstorfer; Jörg F. Rippmann; Sanne Skovgård Veidal; Kristoffer T. G. Rigbolt; Tanja Schönberger; Matthew P. Gillum; Henrik Hansen; Niels Vrang; Jacob Jelsing; et al. Disease Progression and Pharmacological Intervention in a Nutrient-Deficient Rat Model of Nonalcoholic Steatohepatitis. Digestive Diseases and Sciences 2018, 64, 1238-1256, 10.1007/s10620-018-5395-7.
- Kashireddy, P.V.; Rao, M.S. Lack of peroxisome proliferator-activated receptor α in mice enhances methionine and choline deficient diet-induced steatohepatitis. Hepatol. Res. 2004, 30, 104–110. [Google Scholar] [CrossRef]
- Samuele De Minicis; Laura Agostinelli; Chiara Rychlicki; Gian Pio Sorice; Stefania Saccomanno; Cinzia Candelaresi; Andrea Giaccari; Luciano Trozzi; Irene Pierantonelli; Eleonora Mingarelli; et al. HCC Development Is Associated to Peripheral Insulin Resistance in a Mouse Model of NASH. PLOS ONE 2014, 9, e97136, 10.1371/journal.pone.0097136.
- Hu-Quan Yin; Mingoo Kim; Ju-Han Kim; Gu Kong; Mi-Ock Lee; Kyung-Sun Kang; Byung-Il Yoon; Hyung-Lae Kim; Byung-Hoon Lee; Hepatic gene expression profiling and lipid homeostasis in mice exposed to steatogenic drug, tetracycline.. Toxicological Sciences 2006, 94, , 10.1093/toxsci/kfl078.
- Sayantan Nath; Sankar Kumar Ghosh; Yashmin Choudhury; A murine model of type 2 diabetes mellitus developed using a combination of high fat diet and multiple low doses of streptozotocin treatment mimics the metabolic characteristics of type 2 diabetes mellitus in humans. Journal of Pharmacological and Toxicological Methods 2017, 84, 20-30, 10.1016/j.vascn.2016.10.007.
- Martijn W H Leenders; Maarten W Nijkamp; Inne H M Borel Rinkes; Mouse models in liver cancer research: a review of current literature.. World Journal of Gastroenterology 2008, 14, , .
- Dianne H. Dapito; Ali Mencin; Geum-Youn Gwak; Jean-Philippe Pradère; Myoung-Kuk Jang; Ingmar Mederacke; Jorge M. Caviglia; Hossein Khiabanian; Adebowale Adeyemi; Ramon Bataller; et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell 2012, 21, 504-16, 10.1016/j.ccr.2012.02.007.
- Michelle Dow; Rachel M. Pyke; Brian Y. Tsui; Ludmil B. Alexandrov; Hayato Nakagawa; Koji Taniguchi; Ekihiro Seki; Olivier Harismendy; Shabnam Shalapour; Michael Karin; et al. Integrative genomic analysis of mouse and human hepatocellular carcinoma. Proceedings of the National Academy of Sciences 2018, 115, E9879-E9888, 10.1073/pnas.1811029115.
- Ju-Seog Lee; In-Sun Chu; Arsen Mikaelyan; Diego F Calvisi; Jeonghoon Heo; Janardan K Reddy; Snorri S Thorgeirsson; Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nature Genetics 2004, 36, 1306-1311, 10.1038/ng1481.
- Pimpin, L.; Cortez-Pinto, H.; Negro, F.; Corbould, E.; Lazarus, J.V.; Webber, L.; Sheron, N. EASL HEPAHEALTH Steering Committee Burden of liver disease in Europe: Epidemiology and analysis of risk factors to identify prevention policies. J. Hepatol. 2018, 69, 718–735. [Google Scholar] [CrossRef]
- Thomas Reiberger; Mattias Mandorfer; Beta adrenergic blockade and decompensated cirrhosis. Journal of Hepatology 2017, 66, 849-859, 10.1016/j.jhep.2016.11.001.
- Directive 2010/63/EU of the European Parliament and of the Council on the protection of animals used for scientific purposes. Official Journal of the European Union. 2010. L276/33–L276/79 (22 September 2010). Available online: http://eur-lex.europa.eu/LexUriServ (accessed on 5 November 2019).
- EARA Statement supporting European Directive 2010/63/EU on the protection of animals used for scientific purposes n.d. Available online: http://www.ehnheart.org/projects/1037:eara-statement-supporting-european-directive-201063eu-on-the-protection-of-animals-used-for-scient (accessed on 5 November 2019).
- FELASA working group on revision of guidelines for health monitoring of rodents and rabbits; M Mähler (Convenor); M Berard; R Feinstein; A Gallagher; B Illgen-Wilcke; K Pritchett-Corning; Marcello Raspa; FELASA recommendations for the health monitoring of mouse, rat, hamster, guinea pig and rabbit colonies in breeding and experimental units. Laboratory Animals 2014, 48, 178-192, 10.1177/0023677213516312.
- Russell, W.M.S.; Burch, R. The Principles of Humane Experimental Technique; Methuen: London, UK, 1959. [Google Scholar]
- Sara Gargiulo; Adelaide Greco; Matteo Gramanzini; Silvia Esposito; Andrea Affuso; A. Brunetti; Giancarlo Vesce; Mice Anesthesia, Analgesia, and Care, Part I: Anesthetic Considerations in Preclinical Research. The ILAR Journal 2012, 53, , 10.1093/ilar.53.1.55.