A diet high in phosphorus fed to mice deficient in klotho, a cofactor that regulates phosphate metabolism, accelerates aging, sarcopenia, general organ atrophy, kyphosis, and osteoporosis. Similar effects are seen in phenotypes of mutant p53 mice that overexpress the p53 tumor suppressor gene. Although mutant p53 mice do not develop tumors compared to wild-type mice, mutant p53 mice have shorter mean lifespans. Furthermore, tumorigenesis is associated with the sequestration of excessive inorganic phosphate, and dangerous levels of phosphate are released into circulation during tumor lysis syndrome. In total, this evidence implies that tumorigenesis may be a compensatory mechanism that provides protective effects against systemic exposure to dysregulated phosphate metabolism and phosphate toxicity related to cachexia in cancer.
1. Introduction
The metabolism of the essential dietary mineral phosphorus in the form of inorganic phosphate (Pi) is regulated in the human body by a sensitive network of hormones from bone, kidneys, intestines, and parathyroid glands [
1]. Fibroblast growth factor 23 (FGF23) from bone, and parathyroid hormone (PTH) from the parathyroid glands, downregulate high Pi serum levels by decreasing renal Pi reabsorption and increasing phosphaturia. Conversely, the kidneys produce bioactive vitamin D (calcitriol), which increases Pi intestinal absorption and upregulates Pi serum levels. Overburdening this Pi regulatory network may produce phosphate toxicity, a condition in which high amounts of dysregulated phosphate accumulate in the body and damage most major organ systems. In animal studies, high dietary phosphate intake is associated with stimulated cell signaling through the phosphoinositide 3-kinase/AKT/mTOR pathway, leading to cancer growth as excessive Pi is sequestered into cells and incorporated into the sugar–phosphate backbone of nucleic acids during tumorigenesis [
2].
A recent analysis of data from the U.S. National Health and Nutrition Survey (NHANES) found that cancer was inversely associated with serum levels of klotho, a transmembrane protein produced in kidneys and the brain that functions as a cofactor with FGF23 to downregulate serum Pi [
3]. The forced expression of klotho in laboratory samples of cancer cells reduced cancer cell proliferation, while klotho silencing increased cancer cell proliferation [
4], suggesting that klotho acts as a tumor suppressor in vitro. However, more research is needed to determine whether klotho’s association with tumor suppression is mediated by klotho’s role in downregulating Pi concentrations in cancer cells in vivo. Furthermore, reduced klotho levels in laboratory mice are associated with premature aging [
5]. Of relevance, phosphate toxicity accelerates aging effects in laboratory mice [
6], inferring that the downregulation of dysregulated phosphate by klotho may also mediate the association of klotho with “putative age-suppressing” properties [
7]. In both cancer and aging, the reduction in phosphate toxicity potentially mediates klotho as both a tumor-suppressing agent and as an age-suppressing agent.
Muscle wasting in aging—sarcopenia—shares common pathways with muscle wasting in cancer—cachexia—and “more research should be devoted to the understanding of muscle wasting mediators, both in cancer and ageing” [
8]. “For the practicing oncologist, early identification and management of cachexia is critical” [
9]. Cachexia occurs in up to 80% of advanced cancer patients and directly causes 30% of cancer deaths [
10], but effective treatments for cancer cachexia are still undergoing development. For example, a systematic review of pharmacotherapies for cachexia found promising benefits in clinical trials for anamorelin, a selective ghrelin receptor agonist [
11]. Nevertheless, efforts to treat cancer cachexia with medical or nutritional interventions have failed in the past, and new multimodality approaches are needed to target “mechanisms contributing to cachexia” [
12].
2. Klotho Knockout Mice and p53 Mutant Mice
2.1. Klotho Knockout Mice
The mutated klotho gene and its effect on aging was introduced in a mouse model by Kuro-o et al. in 1997 [
5]. In 2010, Ohnishi and Razzaque [
6] used a klotho knockout mouse model with an inactivated klotho gene to investigate the role of dietary phosphate feeding and phosphate toxicity in accelerating aging effects. Compared to wild-type mice, klotho mutant mice had higher serum phosphate levels associated with increased expression of the protein for sodium–phosphate cotransporters (NaPi2a) within the kidney nephrons. The results of experiments that altered genetic and dietary factors in klotho knockout mice were summarized by researchers as follows:
“Klotho knockout mice (klotho−/−) have a short life span and show numerous physical, biochemical, and morphological features consistent with premature aging, including kyphosis, uncoordinated movement, hypogonadism, infertility, severe skeletal muscle wasting, emphysema, and osteopenia, as well as generalized atrophy of the skin, intestine, thymus, and spleen”.
All the above features in klotho knockout mice are associated with phosphate toxicity from feeding high dietary phosphate, as demonstrated by the researchers through genetic manipulation of sodium–phosphate cotransporters.
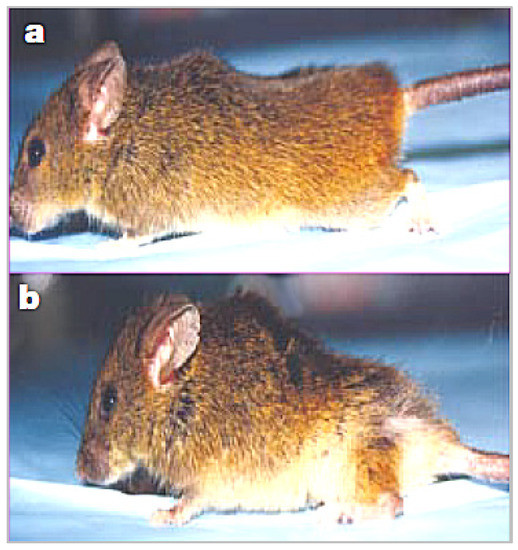
Figure 1, from a study of wound healing in a klotho mutant mouse model, compares a wild-type mouse (a) to a smaller-sized klotho mutant mouse with kyphosis (b) [
17].
2.2. p53 Mutant Mice
The activation of the p53 protein is linked to cellular senescence, “an irreversible process of growth arrest occurring in response to cellular aging” which also serves as a tumor suppression mechanism [
18]. In 2002, Tyner et al. reported generating a p53 mouse with a genetically altered ‘m’ allele that augments tumor suppression (
p53+/m) [
13]. Comparisons with wild-type mice (
p53+/+) include the following findings:
“Skinned, older p53+/m mice exhibit clear reductions in body mass, adipose tissue deposition, muscle mass and a pronounced lordokyphosis”.
The mean mass of quadricep muscles in mutant p53 mice was 2.5-fold lower than in wild-type mice. Although over 45% of wild-type mice developed tumors, the mutant p53 mice “were highly resistant to spontaneous tumours” [
13]. Nevertheless, contrary to expectations, mutant p53 mice had a shorter mean lifespan of 96 weeks compared to 118 weeks in wild-type mice. Additionally, osteoporosis and atrophy of skin, spleen, and other organs occurred in mutant p53 mice.
Figure 2 compares a wild-type mouse from the study (a) with a smaller-sized p53 mutant mouse with kyphosis (b).
This entry is adapted from the peer-reviewed paper 10.3390/metabo12121284