Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Long non-coding RNAs (lncRNAs) are novel genetic biomarkers that can be used as exclusionary tools specific to NDDs. These historical biomarkers have been there for years, so a change in the approach is necessary to better diagnose and treat these NDDs.
- long non-coding (Lnc) RNAs
- neurodegenerative diseases (NDDs)
- Alzheimer’s disease (AD)
1. Long Non-Coding RNAs: An Overview
Long non-coding RNAs, classified as non-coding RNA composed of more than 200 nucleotides, have been highlighted as important biomolecules in the cellular mechanisms responsible for normal development to disease progression. LncRNAs, being just one of the types of non-coding RNA, can be further divided into multiple categories by their genomic position. These categories include intergenic lncRNAs, antisense lncRNAs, intronic lncRNAs, and bidirectional lncRNAs [17]. Along with varying genomic position, the localization of lncRNAs can also differ with studies showing nuclear and cytoplasmic localization [18,19]. The primary mechanism that drives nuclear localization is unknown, but it can potentially be attributed to the recruitment of nuclear retention factors facilitated by sequence motifs [20,21]. When these sequences were removed, lncRNA exportation was favorable, and cytoplasmic localization was possible. Compared to mRNA, the structure and biogenesis of lncRNA are similar due to the addition of poly-A tails, 5′ 7-methylguanoasine capping, and transcription by RNA Polymerase II [22]. Unlike mRNA, however, lncRNAs are relatively less abundant, lack the open reading frame typically found on mRNA, and, most importantly, lack a protein-coding ability [22]. Previously, this last feature of lncRNA, along with other non-coding RNA, led to their classification as “junk” DNA with no discernible function. This has been refuted by recent research showing the roles that lncRNAs plays in various biological processes, such as the progression of cancer, neurodegenerative diseases, and normal development [23,24,25]. It has been shown that lncRNAs play a crucial role in the regulation of diseases course and normal cellular processes through gene regulation. Figure 2 shows the schematic representation of lncRNA function.
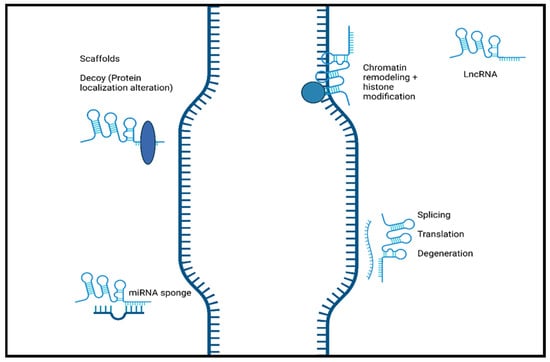
Figure 2. The schematic representation of the function of long non-coding RNA. LncRNAs induce chromatin remodeling and histone modification. Interaction with mRNA. LncRNA hybridization may lead to alternatively spliced transcripts, translation, and mRNA degeneration. LncRNAs interact with proteins/biological molecules to modulate their activity by binding to specific proteins and altering protein localization. They also serve as scaffolds to allow the formation of miRNA sponges. Created with BioRender.com.
Additionally, lncRNA localization correlates highly to function, with nuclear lncRNA functioning primarily as chromatin regulators, while cytoplasmic lncRNAs are more focused on post-translational regulation and the stability of mRNA [21]. LncRNAs have the potential to modulate normal and abnormal development through their mechanisms as decoys, scaffolds, and guides, in addition to direct interaction with DNA and protein molecules [26].
2. LncRNA Mechanisms
LncRNAs play a vital role in disease progression and development that is highly dependent on their well-known functions, including altering gene expression by modulating chromatin structure and regulating transcription and post-transcriptional modifications [27]. These processes can be achieved by lncRNAs acting as guides, decoys, and scaffolds [26,28]. LncRNAs can function as guides and recruit chromatin-remodeling complexes to a specific locus in cis or trans sites [29]. Through recruitment, lncRNAs can target specific sections to alter the structure of chromatin and, therefore, alter the gene expression of that specific gene. LncRNAs can also directly interact with DNA to form hybrid structures such as R-loops or triple helices (triplexes). Triple helices have the potential to mediate gene activation or repression by recruiting remodeling complexes [30]. R-loops can be recognized by a variety of proteins to also promote or suppress gene expression [31]. Furthermore, lncRNAs can interact with proteins and function as scaffolds. As a scaffold, they can aid in the binding of proteins to assemble a complex commonly known as ribonucleoprotein complexes [32]. These complexes have roles in mRNA splicing, translation, and stability, allowing lncRNAs to affect these processes indirectly [18]. LncRNAs can also function as decoys and lower the availability of regulation factors, which can negatively impact gene expression [33]. Furthermore, lncRNAs can regulate pathways and expression through miRNA sponging. This mechanism, enables lncRNAs to act as an miRNA target and bind to miRNA. The result of miRNA sponging is a reduction in the miRNA function, leading to the alteration of signaling pathways that can affect further gene expression [34].
3. LncRNAs in Neurodegenerative Diseases
3.1. Alzheimer’s Disease
Alzheimer’s disease (AD) is a progressive and irreversible neurodegenerative disorder that is the most common form of dementia. This disease is named after Dr. Alois Alzheimer, who noticed that there had been significant changes in the brain of a woman who had unusual symptoms such as memory loss, language problems, and unpredictable behavior. These significant changes are now known as two hallmark characteristics of AD. The first is amyloid plaques, which are produced from amyloid β (Aβ) aggregation, and the second is neurofibrillary tangles (NFT), which are produced from an accumulation of pathological tau [35]. Late-onset AD (LOAD) usually presents as an age-related disease in individuals 65 and over. There is a small portion of individuals with AD who have early-onset Alzheimer’s disease (EOAD), which occurs in people younger than 65 with a genetic predisposition. In the hundred or so years since the discovery of the disease, there have been many studies looking to elucidate the mechanisms of the disease. Although treatments may help relieve some of the symptoms and/or progression associated with NDDs, there are currently no known cures. Therefore, there is a critical need to expand the understanding of what causes neurodegeneration and to develop new approaches for the prevention and treatment of AD [35].
In recent studies, lncRNAs have been shown to be involved in regulatory mechanisms such as transcriptional, posttranscriptional, and translational regulation, as well as a variety of biological functions such as development, differentiation, and metabolism [36]. LncRNAs have also been shown to be involved in neurological diseases such as epilepsy, neurodegenerative diseases, and genetic disorders [37,38,39]. LncRNAs have also been shown to play a role in the pathogenesis of AD, yet the exact mechanism is still unknown. In a microarray analysis study of lncRNAs expressed in the hippocampal region of AD, 315 lncRNAs were found to be dysregulated [40]. Many protein-coding mRNAs have antisense transcript “partners,” which are commonly noncoding RNAs. An example would be the antiscript BACE1-AS, which has been shown to regulate BACE1 mRNA and its protein expression. BACE1 (beta-site amyloid precursor protein cleaving enzyme1) is required for the formation of all monomeric Aβ (1-42) and is also thought to be the cause of toxicity in AD patients [41]. This study shows that lncRNAs are responsible for the increase in Aβ 1-42 in AD, showing that lncRNAs influence the pathogenesis of AD [42].
3.1.1. Competitive Endogenous RNA (ceRNA) Theory
Several studies have reported that lncRNA competes with miRNA target genes by sharing common binding sites [38]. Based on the ceRNA theory, Wang and collaborators created a global triple network where lncRNAs and mRNAs form a triplicate that shares the same miRNA. Based on this network, an AD NFT-associated lncRNA-mRNA network (NFTLMN) was created, mapped, and analyzed, providing three lncRNAs highly related to AD NFT [38]. Gene ontology (GO) function and KEGG pathway enrichment analyses were performed on AP000265.1, KB-1460A1.5, and RP11-145M9.4, showing GO terms for formation and development of the neural tube, neural crest cells, and epithelial tube morphogenesis. Phosphorylation terms were also found during the analysis [38]. A different study found 40 pairs of lncRNAs that shared more than one disordered miRNA, 9 of them correlated with other neurodegenerative disorders, and 5 lncRNAs that could be potential biomarkers for [43]. These machine learning studies have found several lncRNAs that would be suitable for further research as possible biomarkers since they have been identified as showing a correlation with AD genes, but their exact function is unknown.
3.1.2. LncRNA Involvement
Many lncRNAs have been identified as having a role in AD. Although many of these newly identified lncRNAs have been found through bioinformatics, many of these lncRNA functions remain obscure. The lncRNAs related to AD that have been studied have been discovered to be involved in synaptic and neuron exhaustion, neurotrophin depletion, inflammation, mitochondrial impairment, oxidative stress, and DNA damage [44]. lncRNAs, such as NDM29, BC200, 51A, and BACE1-AS, are differentially expressed in AD and correlated with AD progression [45]. Neuroblastoma differentiation marker 29 (NDM29), a lncRNA that promotes the cleavage of BACE and γ-secretase, plays a critical role in AD pathogenesis by inducing an inflammatory response. Studies show NDM29 can induce APP synthesis, increasing Aβ and Aβ-42/Aβ-40 ratio [44,45]. The BACE1 antisense transcript (BACE1-AS) regulates BACE1 mRNA and protein expression when exposed to Aβ-42 [44]. BACE1-AS is significantly upregulated in the cerebellum, hippocampus, entorhinal cortex, and superior frontal gyrus of the AD brain. There is a synergistic mechanism in how BACE1-AS regulates BACE1, which can promote target mRNA or inhibit miRNA [46]. MALAT1 is a highly abundant and evolutionarily conserved lncRNA and regulates a subset of genes involved in synaptic plasticity. Recent studies have reported reduced lncRNA MALAT1 levels in the central spine fluid and brains AD patients compared with a control group [47,48]. Several preclinical studies have provided evidence and support for the potential roles of lncRNA MALAT1 in AD pathogenesis. For instance, studies in experimental AD models indicated enhanced neurite outgrowth, reduced proinflammatory cytokines, decreased neuronal apoptosis, and increased presynaptic bouton density on dendrites with lncRNA MALAT1 overexpression, and vice versa with lncRNA MALAT1 knockdown [49]. Similarly, a recent study suggested the beneficial effect of lncRNA MALAT1 against Aβ1-42-induced toxicity [47]. lncRNA-51A overlaps with SORL1 antisense and could affect amyloid beta formation, which is known to be upregulated in AD [38]. Sortilin-related receptor 1 (SORL1) is one of the genes involved in the processing of amyloid-β protein precursor (APP) and is thought to be a genetic factor of AD. LncRNA-51A downregulates SORL1, which results in abnormal APP processing [50]. BC200 exhibits abnormal subcellular localization and expression levels in specific brain regions in AD patients [37]. BC200 is a protomer-associated RNA that works on the translation level by increasing synapse loss [44]. The loss of synapses is one of the pathological features of AD and is the cause of memory loss in AD patients. In normal aging, BC200 was found to be downregulated, but in AD brains it was found to be significantly upregulated in brain areas related to AD. Furthermore, relative levels of BC200 RNA in AD-affected areas of the brain increased according to the severity of the disease [51]. Li et al., found BC200 could be a positive regulator of BACE1 in AD [52]. Many studies have shown that lncRNA nuclear paraspeckle assembly transport 1 (NEAT1) promotes inflammation and has a role in neurodegenerative disorders. NEAT1 is upregulated in AD and progresses via the miR-124–BACE1 axis showing it can be manipulated to modulate BACE1 expression. This information demonstrates that NEAT1 can be a target for pharmacological therapies and a biomarker for the disease [53]. With the continuing bioinformatic research, several lncRNAs have been identified with tentative functions and correlations. A study by Nana Ma and collaborators identified 487 significantly dysregulated lncRNAs in AD model mice (APP/PS1) brains. These lncRNAs were found to be involved in synaptic plasticity and memory (Akap5), and regulation of amyloid-β induced neuroinflammation (Klf4) [54]. Transcriptomic analysis identified RP3-522J7, MIR3180-2, and MIR3180-3 as lncRNAs which were most highly co-expressed with known AD-related genes [55]. Shi et al., also investigated genomic localization and found that several lncRNAs are located near important protein-coding genes (PCGs) in the human genome. For example, six differentially expressed lncRNAs were within 10 MB of the PCG S100B [55]. These non-coding RNAs have been identified by machine learning and, as such, have no identified function at this time. Figure 3 shows the lncRNAs involved in Alzheimer’s disease.
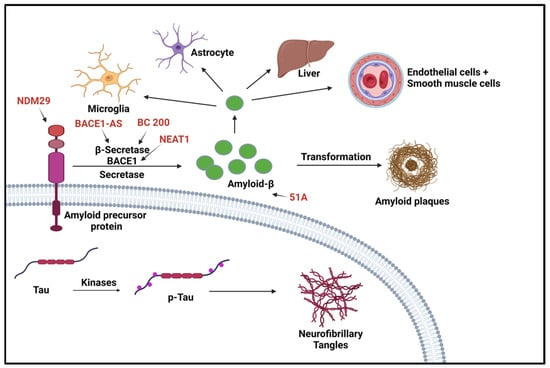
Figure 3. LncRNAs involved in Alzheimer’s disease. Two aspects of this disease are shown here. The first aspect is the production/aggregation of Amyloid-β (Aβ) peptides, which are the final product of amyloid precursor proteins (APPs); the other aspect is the accumulation of neurofibrillary tangles (NFTs) caused by hyperphosphorylated microtubule-associated protein (p-Tau). BACE1 is the rate-limiting enzyme for amyloid precursor proteolysis and is affected by BC200 and BAEC1-AS. NDM29 induces the formation of APP, which in turn promotes an increase in Aβ formation [11]. “Created with BioRender.com”.
3.2. Parkinson’s Disease
Parkinson’s disease (PD) is a progressive neurodegenerative disease that is characterized by death or malfunction of dopaminergic neurons in the substantia nigra and dopamine depletion in the striatum resulting in the loss of motor and non-motor functions. Approximately 60,000 new PD cases are diagnosed each year, joining the 1 million Americans who currently have PD. The direct and indirect annual medical costs for PD approach $25.4 billion and $26.5 billion, respectively. Despite having gained considerable knowledge about the pathological mechanisms of PD over the last several decades, there is little know about how to stop or delay the ongoing neurodegenerative processes. Symptoms typically occur when approximately 70% of the neurons are lost, and other characterizations, such as Lewy bodies composed of α-synuclein, can accumulate in the substantia nigra [56]. Currently, there is no effective cure for PD, and most medications are prescribed for symptom management. Methods of increasing striatal dopamine, such as levodopa, dopamine antagonists, and monoamine oxidase B inhibitors, are used to treat the motor implications of Parkinson’s disease [57]. Although these medications help alleviate motor symptoms, they do not aid in slowing disease progression. Thus, PD patients are in urgent need of disease-modifying therapies that can slow or stop the progression of the neurodegenerative process. The exact molecular mechanisms of PD development are not known. However, lncRNAs have been highly regarded as potential regulators for Parkinson’s disease. This potential is due to their role in biological processes and studies showing altered pathways in PD. Specific lncRNAs have been identified as altering expressions in PD patients compared to controls in samples including brain tissue, blood, and cerebrospinal fluid. Some examples of these lncRNAs include AL049437 and AK021630, which were found to be significantly upregulated and downregulated, respectively, in PD [58]. Mechanistically, lncRNAs found to be involved in PD pathogenicity aid or worsen the disease through various pathways, with most studies targeting primary characteristics in the pathophysiology of PD such as neuronal injury, inflammation, and α-synuclein accumulation.
LncRNA Involvement
Neuronal damage is prevalent in Parkinson’s disease, with most symptoms resulting from the loss of dopaminergic neurons. Some lncRNAs are involved in regulating injury through autophagy and the apoptosis of neuronal cells. LncRNA NEAT1 has been found to be upregulated in mice with MPTP-induced PD, which promotes the stability of PINK1 expression. As a result, it promotes the autophagy caused by MPTP to induce PD [59]. Alternatively, NEAT1 can regulate PD progression in MPP+ SK-N-SH cells by functioning as an miRNA sponge for miR-212-3p and, therefore, modulating the expression of AXIN1 protein to mediate cellular apoptosis [60]. Similarly, lncRNA BDNF-AS, when upregulated, works by downregulating miR-125b-5p to regulate autophagy and apoptosis in cells treated with MPP+ [61]. H19 is also involved in autophagy regulation, where MPP+-induced apoptosis was reduced in H19 overexpressing cells through the negative regulation of miR-585-3p [62]. Additionally, H19 overexpression in induced PD can inhibit the function of miR-301b-3p. When inhibited, it increases the expression of HRPT, usually deficient in PD, through the Wnt/B-catenin pathway, decreasing the loss of dopaminergic neurons [63]. LncRNA Xist is also involved in increasing neuronal injury through the repression of miR-199a-3p. This act allows Xist to induce the expression of transcription factor Sp1, which promotes LRRK2 and has been shown to advance PD progression [64]. Furthermore, lncRNA HOTAIR, which is found to be upregulated, furthered PD through an increase in ROS generation and neuroinflammation; therefore, inducing neuronal injury. HOTAIR potentially increases neuronal injury by regulating the autophagy protein ATG10. The protein expression is promoted through HOTAIR, acting as a sponge for miR-874-5p [65]. Other lncRNA studies have focused on the role lncRNAs play in significant inflammation in PD cases, often regulated by microglial cells, and how this can lead to the progression of neuron injury [66,67]. Patients with PD tend to have a consistent inflammatory response, which could worsen neuron injury [68]. LncRNA MALAT1 was upregulated in MPTP-treated mice, along with increased expression of pro-inflammatory cytokines through epigenetic regulation. In BV2 cells, MALAT1 epigenetically regulated Nrf2 by binding to EZH2. When the expression of Nrf2 is reduced, it increases ROS and inflammation leading to the injury of neurons [69]. LncRNA TUG1 was upregulated in PD-induced mice along with pro-inflammatory cytokines IL-6 and TNF-α [70]. LncRNA UCA1 influences oxidative stress and the expression of TNF-α, IL-6, and IL-1β. In the 6-OHDA PD mouse model, a downregulated UCA1 reduced activation of the PI3K/AKT pathway. The PI3K/AKT pathway is usually involved in a variety of cellular pathways, including neurodegenerative diseases such as PD [71]. Dysregulation of α-synuclein, commonly found in PD, has been correlated to impairment in a variety of cellular processes, such as synaptic vesicles, mitochondria function, and the autophagy-lysosomal pathway, leading to a problem in dopamine levels [72]. These factors make it a therapeutic target for studies determining the involvement of lncRNA regulation and improving PD pathogenesis. LncRNA SNHG1 overexpression reduced miR-15-5p expression and promoted α-synuclein accumulation through the SIAH1 protein. Furthermore, overexpression of lncRNA OIP5-AS1 reduced the α-synuclein accumulation by miR-126 binding and PLK2/α-synuclein autophagy [73]. In a rotenone-induced PD mouse model, lncRNA SNHG14 was discovered to be upregulated with a reversed expression of miR-133b. Upregulation of SNHG14 correlated with increased neuronal injury and an increased a-syn expression through the downregulation of miR-133b [74]. In addition, lncRNA-T199678 expression, through binding and regulation of miR-101-3p, can regulate ROS and apoptosis that was induced by α-syn [75]. Figure 4 shows the lncRNAs involved in Parkinson’s disease.
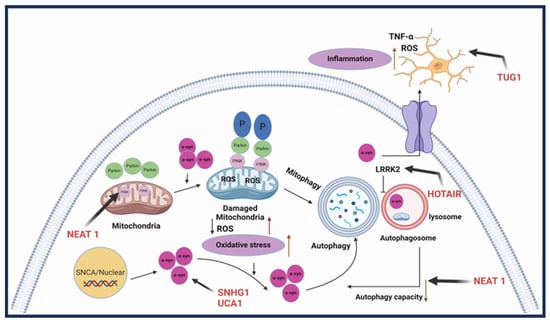
Figure 4. LncRNAs involved in Parkinson’s disease. Parkinson’s disease pathogenesis involves three aspects: the first is dopaminergic neuron death; the second is an aggregation of synuclein-alpha, which forms Lewy bodies; and the final one is neuroinflammation, causing cell death. LncRNA HOTAIR, SNHG1, and UCHL1-AS participate in the accumulation of SNCA. LncRNA NEAT1 causes abnormal autophagy. HOTAIR can also upregulate LRRK2 and thus affect autophagy. LncRNA TUG1 regulates microglia polarization and increases inflammatory cytokine production [11]. Created with BioRender.com.
3.3. Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis, also known as Lou Gehrig’s disease, is a progressive neuromuscular degeneration that primarily affects motor neurons of the somatic nervous system. The gradual erosion of these neuromuscular connections, often starting in distal muscles, encompasses loss in much of the motor functions necessary for daily tasks [76]. Deterioration of the efferent pathways from the brain and spinal cord to effectors incites voluntary muscular atrophy. Though some variation exists in the age of onset, epidemiological studies have shown a positive correlation between age and the number of ALS cases, with the average age of diagnosis being between 55 and 65.1 [77]. Although a generally rare neurodegenerative disorder, ALS has an incidence rate of 2 in 100,000 people annually but increases for older individuals [76,77,78]. Approximately 10–15% of cases result in dual diagnoses of ALS and concomitant frontotemporal dementia (FTD), which shows a wide spectrum of overlapping symptoms and genetics [14,76]. The key differences between ALS and FTD are the targeted locations of deterioration.
Degeneration of the frontal and temporal lobes often corresponds with a range of behavioral changes, whereas ALS is defined by the deterioration of upper and lower motor neurons of the motor cortex that subsequently result in muscle paralysis and atrophy [14]. ALS is known to present itself sporadically (sALS) or genetically. Familial ALS (fALS) constitutes 5–10% of overall cases, where a single allele from a myriad of disease-causing genes will suffice for its onset due to its autosomal dominant pattern of inheritance [76,79]. Unfortunately, individuals suffering from ALS are projected to live 2–5 years after its onset [80]. Genes notorious for their involvement in the development of ALS include mutated or dysregulated ALS2, NEFH, C9orf72, SOD1, FUS, and TARDBP [2,76,81,82].
While genetic biomarkers can provide an assessment of the risk of ALS development, significant heterogeneity has also been associated with the disease, and their exact functions in ALS development and progression are unclear [76,82,83]. Additionally, many other factors are engaged in disease development and progression, including epigenetics, environment, and age-related issues such as oxidative stress [2]. Nevertheless, the average patient dies within 2–5 years [76]. Clinicians are currently restrained from providing palliative care with anti-excitatory drugs to improve patient quality of life, as there is no cure for ALS [79]. With a lack of consensus on standard and specific biomarkers, ALS cases continue to pose a grave threat. The severity of late-stage disease and ambiguity of early-stage symptoms of ALS sets grounds to demand more accurate diagnostic and prognostic biomarkers. LncRNAs have been implicated in regulatory mechanisms leading to ALS development and progression and show high tissue specificity, making them prime targets for more effective diagnostics and therapeutics [84].
LncRNA Involvement
Several studies have shown abnormal RNA metabolism from mutagenic RNA-binding proteins FUS, C9orf72, and TDP-43 to be an innate characteristic of ALS pathogenesis [84,85,86]. Paraspeckles are essential components with protective roles in the cellular stress response of motor neurons (MNs). A functional aspect of paraspeckles is their involvement in nucleoplasmic sequestration of RNA and proteins that directly alters target site expression [87]. The aggregation of paraspeckles in the CNS is a hallmark feature of ALS. LncRNA NEAT1 has inherent roles as a scaffold for paraspeckle formation [85,88]. Enriched lncRNA NEAT1_2 is proved to be the target of both FUS and TDP-43 through its UG-rich sequences; however, mutagenic or dysregulated RNA-binding proteins are associated with distorted or hyper-assembly of paraspeckles in early ALS pathogenesis [86,89]. In vivo studies have identified NEAT1 involvement in specific neurodegenerative pathways such as inflammation and neuronal cell death through p53 regulation; hence, the decreased brain density seen in brain scans and postmortem examinations of many NDD patients [81]. Of course, paraspeckles are primarily absent in healthy MNs because of decreased expression of NEAT1 isoforms essential to their assembly in the central nervous system (CNS), as studied with in vitro post-mitotic neurons [85,89]. Furthermore, NEAT1 expression has roles in neuron-specific pathways, and the aberrant hyperexcitability of affected MNs has been implicated in ALS and other NDDs [85]. The sequestering of genetic regulatory elements, including miRNAs by paraspeckle formation through abnormal NEAT1 expression in response to stressful external stimuli, such as proteasomal inhibition and viral infections, have also been identified in ALS [90]. These findings confer lncRNA NEAT1 in the CNS to be a viable target for ALS therapeutics.
Other discoveries of ncRNA’s influence in the pathophysiology of NDDs include antisense C9orf72 transcripts that have been connected to chromosome 9p-linked ALS. The C9orf72 antisense transcript appears to be highly conducive in fALS development, with 22.5% of fALS cases attributable to the hexanucleotide repeat expansion [91]. Experimental findings show various disease mechanisms the transcript acts through, though it has a significant role in nuclear RNA sequestration, which directly impacts transcription [92]. LncRNA CCND1, a FUS-bound transcript and cell proliferation regulator, is another ncRNA associated with ALS, particularly through the Wnt/β-catenin signaling pathway [93,94,95]. FUS acts as an inhibitor to CCND1 expression to regulate cell cycle progression; however, its dysregulation may trigger apoptosis in ALS [96]. Figure 5 shows the lncRNAs involved in ALS.
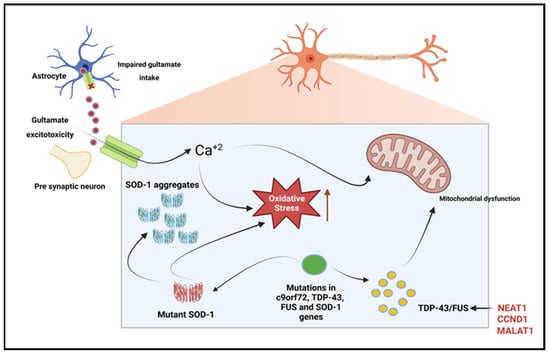
Figure 5. LncRNAs involved in amyotrophic lateral sclerosis. Due to dysfunction of the astrocytic excitatory amino acid transporter 2 (EAAT2), there is reduced uptake of glutamate from the synaptic cleft, which leads to glutamate excitotoxicity. The resulting glutamate-induced excitotoxicity induces neurodegeneration through the activation of Ca+2 dependent pathways. TDP-43, c9orf72, and fused in sarcoma (FUS) gene mutations result in dysregulated RNA metabolism, leading to the formation of intracellular neuronal aggregates. Superoxide dismutase-1 (SOD-1) gene mutations increase oxidative stress and induce mitochondrial dysfunction, leading to intracellular aggregates. Enriched lncRNA NEAT1 is proved to be the target of both FUS and TDP-43 [97]. Created with BioRender.com.
Dingsheng et al. generated an ALS-specific competitive endogenous RNA (ceRNA) network to investigate RNA transcripts with sponging effects on miRNAs to differentially regulate their expression in ALS [98]. Notably, MALAT1 was found to act as a sponge to modulate the expression of 75 genes, 7 of which have been connected to the pathogenesis of ALS by interacting with miRNA [98]. Furthermore, MALAT1, like NEAT1, is bound by TDP-43, an RNA-binding protein involved in ALS [99]. TDP-43 is noted to be a causative agent of mitochondrial dysfunction with implications in neuroinflammation, a common feature of early-stage ALS. MALAT1 is also known to regulate hnRNPA2/B1 and XIAP, both of which are closely related to apoptotic pathways that lead to neurodegeneration in ALS [98]. Experimental findings show that MALAT1 regulates the ATM gene, which is a member of the p53 apoptotic pathway in response to the DNA damage linked to the pathogenesis of ALS. Moreover, the endocytic pathway protein AAK1, which associates with SOD1 mutants and has been implicated in ALS, has also been identified as a regulatory target of MALAT1 [98]. Evidently, MALAT1 is another crucial player that can potentially have a role as a therapeutic target and a potential biomarker for ALS assessments or targeted therapies.
While a large pool of research is accessible for coding transcripts, detailed investigations of lncRNA involvement in ALS pathogenesis are in their infancy. Nevertheless, recent literature introduces differential expression of lncRNAs with impacts on transcriptional regulation pathways. ZEB1-AS1, ZBTB11-AS1, and XXbac-BPG252P9.10 are reported as novel antisense transcripts involved in ALS transcriptional regulation [41]. ZEB1-AS1 gets its significance as an antisense transcriptional regulator of Zinc Finger E-Box Binding Homeobox 1 (ZEB1), which is a highly conserved transcriptional repressor with roles in chromatin and E-Box binding; however, ZEB1-AS1 was downregulated in sALS samples in comparison to healthy control groups [98]. ZBTB11-AS1 is another differentially expressed antisense transcript coupled with the Zinc Finger and BTB Domain Counting 11 gene (ZBTB11), implying its participation in transcriptional regulation. Interestingly, cases of sALS presented with downregulated ZBTB11-AS1 [100]. XXbac-BPG252P9.10 is associated with the nuclear factor-kappa-B/REL (NF-κB) transcription factor family. The NF-κB proteins have critical roles in inflammatory and survival pathways but, in particular, IER3 is known for its regulation of anti-apoptotic genes and roles in ALS. LncRNA XXbac-BPG252P9.10 is an antisense regulator of IER3 and is significantly downregulated in sALS samples [101]. Non-coding antisense RNA expression analyses and co-expression networks involving lncRNAs and mRNAs show a multitude of potential transcripts with involvement in ALS pathogenesis that may prove to be promising therapeutic or diagnostic targets. Some relevant lncRNAs and their involvement in different neurodegenerative diseases have been summarized in Table 1.
Table 1. Summary of relevant lncRNAs and their involvement in different neurodegenerative diseases.
| LncRNA | Type of NDD | Notes | References |
|---|---|---|---|
| BACE1-AS | Alzheimer’s | BACE1-AS has been associated with regulation of BACE1, a key enzyme in amyloid β production | [53,102,103] |
| NEAT1 | Parkinson’s | Associated with modulation of neuronal apoptosis and neuro-inflammation | [53,104,105] |
| MALAT1 | Alzheimer’s | Linked with neuronal apoptosis and neuro-inflammation | [48,106,107] |
| SNHG1 | Alzheimer’s/Parkinson’s | Plays a role in regulation of amyloid β production and neuro-inflammation | [108,109,110] |
| ANRIL | Parkinson’s | Associated with vascular dysfunction and inflammation in CNS | [111] |
| HOTAIR | Alzheimer’s | Associated with dysregulation of synaptic plasticity and neuronal apoptosis, contributing to cognitive decline | [112,113] |
| TUG1 | Parkinson’s | Shown to modulate dopaminergic neuronal cell death suggesting its involvement in pathogenesis of Parkinson’s | [70,114] |
| BC200 | Parkinson’s/Multiple sclerosis | Involved in regulating mRNA translation and synaptic plasticity and contributing to disease progression | [115,116] |
| MEG3 | Alzheimer’s | Implicated in amyloid β-induced neurotoxicity and neuronal apoptosis | [117] |
| PINK-AS | Parkinson’s | Impairment of mitochondrial dynamics due to decrease in the PINK1-AS and neurodegeneration due to ASUCHL1 downregulation | [118] |
| NDM29 | Alzheimer’s | NDM29 expression is enhanced in the cerebral cortex of AD patients | [45,119] |
| LRP1-AS | Alzheimer’s | LRP1 is deeply involved in APP trafficking and Aβ processing | [120,121] |
This entry is adapted from the peer-reviewed paper 10.3390/ijms25042268
This entry is offline, you can click here to edit this entry!