1. Introduction
Heat stress refers to physiological responses occurring due to prolonged exposures to high temperatures, often combined with elevated humidity, that cause body heat gain to exceed one’s ability to dissipate the heat [
1]. It arises when the body is unable to thermoregulate properly, leading to an abnormal rise in core body temperature [
2]. The fundamental origin of escalating heat stress is global warming stemming from anthropogenic climate change—the accumulation of greenhouse gases like carbon dioxide, methane, and nitrous oxides due to human activities including burning fossil fuels, deforestation, and agriculture [
3]. One key mechanism is through the impairment of evaporative cooling from sweating and respiration [
4]. High humidity in particular hampers heat loss via sweating and skin evaporation by reducing the vapor pressure gradient from the skin to the environment [
5]. Studies show that humid heatwaves have become more frequent worldwide [
6], causing instances of near-fatal and fatal hyperthermia and heat stroke by blocking the primary heat loss avenue in humans [
7]. The multifaceted effects of escalating heat stress can be detrimental with widespread repercussions for health, economies, ecosystems, and more. Some major anticipated impacts include dramatic rises in heat-related compromised reproductive efficiency [
8]. The deleterious effects of heat stress (HS) on reproductive function encompass a comprehensive spectrum of repercussions affecting both male and female reproductive components, spanning from the intricate mechanisms of fertilization to the critical stages of early and late embryo–fetal development. Consistently, extensive investigations have collectively unveiled the multifaceted impacts of HS on the reproductive landscape [
9,
10,
11,
12,
13].
Within the sphere of male reproductive physiology, HS emerges as a pivotal factor intricately linked to male infertility, exerting a discernible influence on testicular functionality [
13,
14]. Notably, empirical evidence drawn from diverse studies underscores the detrimental ramifications of HS on male fertility and semen quality [
15,
16,
17,
18,
19,
20,
21,
22]. Furthermore, HS casts a shadow over the ensuing fertilization processes, as elucidated by recent investigations [
23,
24,
25]. Recent scientific inquiries have accentuated the detrimental influence of HS on sperm quality across a spectrum of mammalian species, encompassing dogs [
17], bulls [
20], buffaloes [
26], stallions [
27,
28,
29], rabbits [
30], pigs [
31], goats [
32], and human males [
33,
34]. Remarkably, a recurring theme in these studies underscores the adverse impact of HS-induced oxidative stress and apoptosis on the viability of Sertoli cells, spermatogonial stem cells, Leyding cells, spermatogenesis, and sperm quality [
15,
35,
36,
37,
38,
39,
40,
41,
42]. Furthermore, recent publications have elucidated a compelling association between sperm quality and environmental temperature in men, underscoring the pervasive implications of environmental factors on male reproductive health [
43,
44,
45,
46]. These findings collectively reinforce the imperative need for comprehensive research into mitigating the detrimental effects of HS on reproductive function across species, as well as the exploration of potential interventions to safeguard male fertility and sperm quality under challenging environmental conditions.
In the United States, cattle reproduction and milk production rates experience a pronounced decline during the hot season due to HS, resulting in approximately USD 900 million in annual losses within the dairy industry [
47,
48]. Besides the metabolic stress associated with high milk production in dairy cows, seasonal impacts on fertility have been extensively documented [
49,
50]. Notably, a substantial reduction in cattle fertility during the summer months has been attributed to the thermoregulatory challenges posed by HS, leading to elevated body temperatures [
51,
52,
53]. Furthermore, HS exerts detrimental effects on follicle quality and hormonal equilibrium, contributing to a decline in estrus [
54,
55,
56,
57,
58]. Specifically, granulosa cells responsible for estradiol production are adversely affected by HS, resulting in decreased estradiol production and subsequent disruption of the estrous cycle in cattle [
59,
60,
61]. HS has also been shown to influence corticoid levels, luteinizing hormone (LH), and plasma progesterone in cattle, with alterations in hypothalamus and pituitary gland function leading to changes in the secretion of reproductive hormones [
9,
62,
63,
64,
65,
66]. Moreover, HS negatively impacts oocyte quality due to its effects on the hypothalamus and pituitary gland, specifically the LH [
67,
68,
69]. Additionally, Khan et al. [
47] reported that HS significantly compromises granulosa cells, leading to suboptimal oocyte development. This is further supported by studies that found HS compromises the mRNA expression levels of Moloney sarcoma oncogene (MOS), growth factor 9 (GDF9), and POU domain, class 5, transcription factor 1 (POUF51), which are crucial for oocyte development and competence, ultimately resulting in poor-quality oocytes and impaired embryonic development [
60,
61,
70,
71,
72].
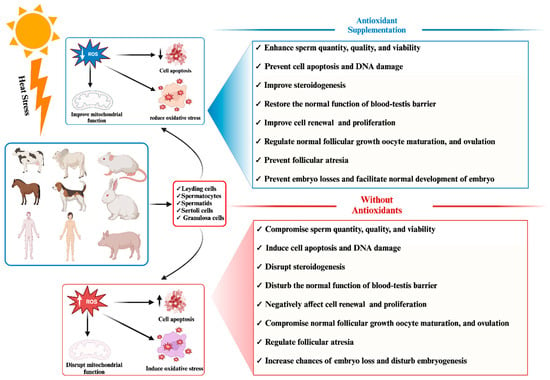
Figure 1 demonstrates the adverse impact of heat stress on phenotypic traits related to mammalian reproductive traits.
Figure 1. Effect of antioxidant supplementation on mammalian reproductive cells under heat stress: pre- and post-supplementation comparison.
In response to these challenges, numerous researchers have advocated for exogenous supplementation with antioxidants to mitigate the elevated levels of reactive oxygen species (ROS) induced by HS and enhance the antioxidant capacity of mammalian reproductive cells [
30,
31,
47,
73,
74,
75,
76]. Consistently, positive outcomes have been observed for supplementation with antioxidants, whether in the form of herbal medicines or feed additives, in improving the efficiency of mammalian reproductive cells such as granulosa cells, Leydig cells, and Sertoli cells by alleviating oxidative stress [
77,
78,
79,
80,
81,
82,
83,
84,
85]. The beneficial impact of antioxidant supplementation in mitigating the adverse effects of heat stress on phenotypic traits related to mammalian reproduction has been succinctly illustrated in
Figure 1.
2. Impact of Heat-Stress-Induced Oxidative Stress and Apoptosis on Mammalian Reproductive Cell Functionality
It is well established that heat stress induces ROS production primarily through mitochondrial dysfunction, where the electron transport chain is compromised, leading to electron leakage and the formation of superoxide radicals (O
−2) [
86,
87]. Additionally, another crucial source of ROS induction during heat stress is the activation of NADPH oxidases (NOXs), particularly NOX2 and NOX4. These enzymes are responsible for generating superoxide ions in response to various stressors, including heat. The activation of NOX enzymes can occur through heat-induced signaling pathways, such as the activation of protein kinase C (PKC) and mitogen-activated protein kinases (MAPKs). The NADPH oxidases also contribute to the production of superoxide radicals during heat stress [
88]. Moreover, these superoxide radicals can be further converted into other types of ROS, such as hydrogen peroxide (H
2O
2) and hydroxyl radicals (OH), through various reactions, including those catalyzed by superoxide dismutase (SOD) [
89,
90]. Consequently, heat-stress-induced ROS can also trigger lipid peroxidation, leading to the formation of lipid peroxidation products such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE). These products are known to be cytotoxic and can disrupt cellular membranes [
90,
91]. These molecules, while playing roles in signaling under normal physiological conditions, can cause significant damage to cellular structures and DNA when present in excess due to heat stress [
90,
91,
92].
Oxidative damage and cell apoptosis represent pivotal consequences affecting mammalian reproductive cells, primarily initiated by the excessive generation of ROS [
93,
94,
95]. The accrual of ROS disrupts male reproductive functions and exerts detrimental effects on semen quality [
96,
97]. These detrimental effects manifest in two main modes: firstly, the overabundance of ROS depletes the cell’s scavenging capacity, impairs antioxidant enzymes, escalates lipid peroxidation, and triggers DNA damage, ultimately compromising the cell’s defense against oxidative harm. Secondly, the surplus ROS mediate molecular signaling in the mitochondria-dependent apoptotic pathway, encompassing events such as the opening of mitochondrial permeability transition pores, mitochondrial membrane depolarization, and the release of mitochondrial substances, including cytochrome C (cyto-c). This, in turn, culminates in caspase-3 activation and subsequent cell apoptosis. Recent research by Li H et al. [
98] further reported that heat stress upregulated caspase-3 and caspase-9, leading to enhanced apoptosis in endometrial epithelial and glandular epithelial cells, along with alterations in HO-1 mRNA/protein and Keap1 mRNA/protein expression, and an elevated malondialdehyde (MDA) level in mouse uterine tissue.
In the context of male reproductive cells, recent investigations have extensively explored the impact of heat stress on testis morphology, antioxidant status, and testicular biosynthesis [
34,
99,
100,
101]. Sertoli cells, known for providing structural and nutritional support for developing germ cells, have been a focus of scrutiny, with studies comprehensively examining the repercussions of heat stress on male reproductive cells, including Sertoli cells, spermatogonial stem cells, and Leydig cells [
102]. Wang C et al. [
103] reported that HS induces oxidative stress and apoptosis in Sertoli cells, disrupting the normal spermatogenesis process. Furthermore, boar Sertoli cells exposed to elevated temperatures exhibited increased oxidative stress and apoptosis, an inhibited pentose phosphate pathway, and decreased ATP content. Molecular changes observed in boar Sertoli cells under heat stress involved the downregulation of the Kelch-like ECH-associated protein 1 (KEAP1)/nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway (associated with enhanced antioxidants) and low levels of heat shock protein 90 (HSP90) due to the suppression of melatonin receptor 1B (MTNR1B), resulting in abnormal regulation of stabilizing hypoxia-inducible factor-1α (HIF-1α) [
104]. Another study by Xue H et al. [
105] found that HS primarily enhanced the lipid oxidation, oxidative stress, and apoptosis in Sertoli cells through the activation of arachidonate 15-lipoxygenase type B (ALOX15B) and the production of 8-hydroxyeicosatetraenoic acid (8-HETE) and 15-hydroxyeicosatetraenoic acid (15-HETE), with involvement of the P53-p38 pathway [
105]. The disruption of arachidonic acid (AA) metabolism, a precursor to 20-carbon polyunsaturated fats, has been reported to be associated with poor spermatogenesis outcomes, as excessive AA levels altered cytomembrane structure and function and increased permeability and brittleness, potentially leading to mitochondrial changes, apoptosis, or necrosis. HS was found to significantly elevate AA levels, disrupting the function of tight junctions (TJs) essential for spermatogenesis development [
106]. Additionally, AA increased MDA levels, activated p38 mitogen-activated protein kinases (P38 MAPKs), and reduced mitochondrial DNA (mtDNA). Furthermore, another study noted that heat stress induced oxidative stress in Sertoli cells by suppressing the level of nuclear factor erythroid 2-related factor 2 (Nrf2) [
107]. In addition, the effects of oxidative stress on Leydig cells have been briefly reviewed in recent studies [
102,
108]. Heat stress treatment inhibited cell viability, induced apoptosis, increased the activity of caspase 3 and the pro-apoptotic protein Bax, and decreased the expression of anti-apoptotic protein B-cell leukemia/lymphoma-2 (Bcl-2), concurrently activating endoplasmic reticulum (ER) stress markers such as glucose-regulated protein 78 (GRP78) and CCAAT/enhancer-binding protein homologous protein (CHOP) [
109].
The impact of heat-stress-induced oxidative stress and apoptosis on mammalian female reproductive cells has also been extensively documented in recent research [
110]. In alignment with these findings, it has been observed that heat-stress-induced oxidative stress and apoptosis in bovine granulosa cells disrupt the normal secretion of estrogen, leading to disturbances in the ovarian microenvironment and subsequent interference with ovarian function [
111]. Consistently, a study documented abnormal folliculogenesis including impaired ovulation, fertilization, and early embryo development [
112]. Additionally, a study has reported elevated levels of ROS production in response to heat stress, resulting in increased apoptosis and even embryo death, coupled with reductions in both mitochondrial activity and membrane potential [
113]. Consistently, Sammad et al. [
114] have reported elevated levels of ROS and apoptosis in bovine granulosa cells under heat stress conditions. They also noted the negative regulation of several candidate genes, including heme oxygenase 1 (HMOX1), nitric oxide synthase 2 (NOS2), catalase (CAT), superoxide dismutase (SOD), B-cell lymphoma 2-like 1 (BCL2L1), glutathione peroxidase 4 (GPX4), Nrf2, aspartoacylase 3 (ASP3), peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PPARGCIA), solute carrier family 16 member 3 (SLC16A3), sterol regulatory element-binding protein 1 (SERBP1), sirtuin 1 (SIRT1), AMP-activated protein kinase (AMPK), Caspase 8 (CASP8), CASP9, insulin-like growth factor 2 (IGF2), peroxisome proliferator-activated receptor alpha (PPARA), and solute carrier family 27 member 3 (SLC27A3), which are associated with apoptosis, cell proliferation, and oxidative activity of granulosa cells [
114]. Furthermore, their research indicated that heat stress significantly downregulated the key anti-apoptotic and antioxidant-associated signaling pathways, including the AMP and Nrf2 signaling pathways [
114]. Similarly, another study reported a significant decrease in the number of primordial follicles, an increase in the number of degenerated follicles, and a decrease in granulosa cell proliferation in response to heat stress [
115]. The molecular mechanisms associated with heat-stress-induced oxidative stress and apoptosis effects on mammalian reproductive cells are summarized in
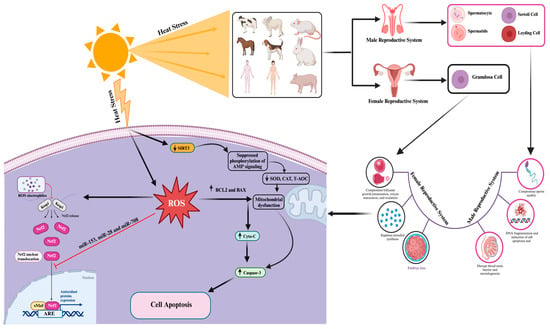
Figure 2 and
Table 1.
Figure 2. Cellular and molecular responses to heat-stress-induced oxidative stress and cell apoptosis in mammalian reproductive cells. The “→” shows direct correlation/effect, while “

” indicates the effect has been suppressed.
Table 1. Effect of heat stress on mammalian reproductive cells.
| Heat Stress |
Biological Effect |
Cells |
Reference |
| |
- ⟡
-
Enhanced accumulation of ROS, suppressed SOD, CAT, and proliferating cell nuclear antigen (PCNA) protein expression levels
|
Sertoli cells |
[116] |
| |
- ⟡
-
Elevates the level of arachidonic acid which disrupts TJs and enhances oxidative stress and apoptosis
- ⟡
-
Disrupts the normal process of spermatogenesis
|
Sertoli cells |
[106] |
| |
- ⟡
-
Elevated the transforming growth factor beta 1 (TGFβ1)/SMAD family member (Smad2)/Smad3 pathway protein expression, causing cell apoptosis, testicular tissue organic lesions, and testicular damage and affecting testicular secretion function.
|
Testis |
[117] |
| |
- ⟡
-
Increased the process of apoptosis by enhancing the level of Bcl-2-associated X protein (BAX) and Caspase-3
- ⟡
-
Suppressed cell proliferation by downregulating PCNA and CyclinB1
- ⟡
-
Enhanced ROS and oxidative stress by suppressing the level of SOD
- ⟡
-
Disrupted the synthesis of progesterone and estrogen by downregulation of the expression of steroidogenic acute regulatory protein (STAR), Cyp11A1
|
Ovarian granulosa cells |
[95] |
| |
- ⟡
-
Induced apoptosis via endoplasmic reticulum stress signaling
- ⟡
-
Upregulates the expression of apoptotic linked genes (caspase-3, BAX, glucose-regulated protein 78 (GRP78) and CHOP) and downregulates BCL2 gene expression
|
Mouse granulosa cells |
[118] |
3. Advancement and Understanding of Genetic Biomarkers Associated with Heat Stress Resistance and Reduced Apoptosis and Oxidative Stress in Mammalian Reproductive Cells
It is well established that heat resistance in mammalian reproductive cells is mediated by a network of genes and their signaling pathways to counteract the damaging effects of heat stress. Based on published data, several genes and pathways, including HSP family genes; anti-apoptotic genes; genes encoding antioxidant enzymes; and the AMPK, ERK1/2, and Nrf2/Keap1 signaling pathways, have been found to be involved in protective mechanisms against heat-stress-induced apoptosis and oxidative stress in mammalian reproductive cells. Detailed information regarding the protective role of the aforementioned genes and pathways against heat stress in mammalian reproductive cells is provided below.
3.1. Role of Heat Shock Protein (HSP) Genes in Mitigating Heat-Stress-Induced Oxidative Stress and Apoptosis in Mammalian Reproductive Cells
The heat shock protein-72 (Hsp72) gene, a prominent member of the heat shock protein (HSP) family, serves as the primary inducible heat shock protein. Its baseline expression in healthy cells is minimal, but it becomes markedly upregulated in response to heat stress. Notably, HSP72 has demonstrated its ability to counteract heat-stress-induced ROS in bovine Sertoli cells when exposed to puerarin, a traditional Chinese medicinal compound [
13]. In this context, HSP72 functions as both an antioxidant and an anti-apoptotic factor within Sertoli cells. It achieves this by reducing ROS production and safeguarding Sertoli cells against oxidative harm and apoptotic processes. Consistently, a study reported the protecting and self-recovering role of HSP70 in bovine oocytes after exposure to severe heat stress [
119]. Furthermore, it revealed that HSP70 prevents apoptosis, supports signal transduction, increases the antioxidant protection of the embryo, as well as protecting heat-stressed maturing bovine oocytes and restoring their developmental competence. Consistently, Ho et al. [
59] observed that Asparagus officinalis stem (EAS) elicited an upregulation of HSP70 and heat shock factor 1 (HSF1) expression, resulting in an augmented concentration of progesterone within heat-treated bovine cumulus–granulosa cells. Additionally, EAS demonstrated the capacity to heighten glutathione (GSH) levels, improve mitochondrial function, and mitigate ROS levels in heat-stressed bovine cumulus–granulosa cells. Notably, when HSP70 was inhibited by Ho et al., a subsequent decrease was noted in the levels of progesterone, GSH, HSF1, Nrf2, and Kelch-like ECH-associated protein 1 (Keap1). These findings collectively underscore the pivotal role of HSP70 as the principal regulator orchestrating antioxidant activity, thereby safeguarding granulosa cells against the deleterious effects of heat stress [
59].
Heme oxygenase 1 (HO-1), alternatively known as heat shock protein-32 (Hsp32), is a stress-responsive enzyme with pivotal roles in maintaining iron homeostasis, fortifying antioxidant defenses, and averting apoptosis [
120,
121,
122]. Interestingly, studies have disclosed an association between low serum levels of HO-1 and an elevated risk of polycystic ovarian syndrome [
123]. Furthermore, investigations have substantiated that HO-1 attenuates heat-stress-induced apoptosis in bovine granulosa cells by curbing ROS production and activating antioxidant responses [
124]. Remarkably, HO-1 modulation influences apoptotic processes, with its downregulation intensifying apoptosis and its upregulation mitigating apoptosis through the regulation of Bax/Bcl-2 expression and cleaved caspase-3 levels [
111]. Additionally, HO-1 plays a cytoprotective role by influencing estrogen levels and catalyzing the breakdown of heme to generate biologically active carbon monoxide (CO). Significantly, CO elevation coincides with heightened HO-1 levels, diminished Bax/Bcl-2 ratios, and inhibition of the extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathway (
Figure 2) [
111]. Studies have shown that reducing antioxidant gene levels in heat-stressed (40 °C) HO-1-knockdown bovine granulosa cells leads to increased cellular apoptosis [
124]. Moreover, research has elucidated that heat stress triggers the activation of Nrf2, which safeguards bovine granulosa cells from heat-stress-induced apoptosis by regulating HO-1. This, in turn, modulates ROS levels, reducing their production and subsequently suppressing oxidative stress and apoptosis [
124].
3.2. Protective Role of SOD Genes against Heat-Stress-Induced Oxidative Stress and Apoptosis in Mammalian Reproductive Cells
The SOD genes’ protective role against heat stress has been established in mammalian reproductive cells [
95,
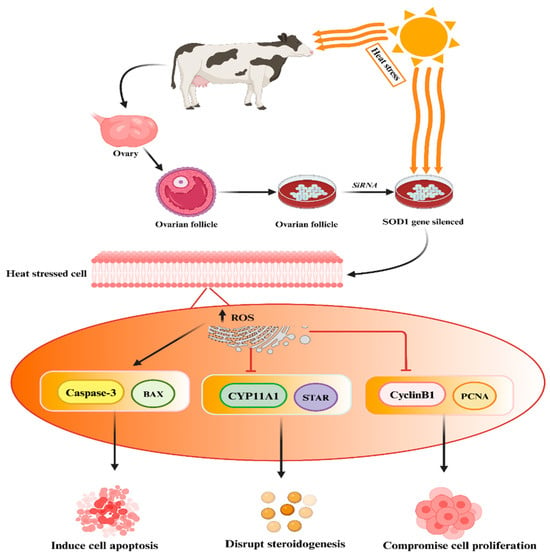
125]. Khan et al. conducted experimental studies demonstrating that the silencing of the SOD1 gene in heat-treated granulosa cells resulted in increased apoptosis, reduced cell proliferation, and decreased biosynthesis of estrogen and progesterone hormones, as depicted in the accompanying
Figure 3 [
95]. In addition, Faheem et al. conducted a study where they observed that under conditions of heat stress, buffalo granulosa cells demonstrated elevated expression levels of SOD2 and an enhancement in total antioxidant activity [
126]. Furthermore, Faheem et al. highlighted the enhanced antioxidant capacity and cholesterol levels in granulosa cells, which likely contribute significantly to their biological function in preventing heat-stress-induced apoptosis and oxidative stress [
125]. These findings suggest that SOD1 plays a key role in regulating other genes while protecting buffalo granulosa cells from the adverse effects of heat stress.
Figure 3. Silencing of SOD1 genes may lead to elevated levels of ROS followed by disruption of steroidogenesis, compromised cell proliferation, and increased cell apoptosis. The “→” shows direct correlation/effect, while “
” indicates the effect has been suppressed.
3.3. ERK1/2 Signaling Pathway Protects Mammalian Reproductive Cells from Heat-Stress-Induced Apoptosis
The ERK1/2 kinases are highly conserved serine–threonine kinases with widespread distribution, playing a pivotal role in cellular signaling regulation, both in normal physiological conditions and pathological states, by phosphorylating various substrates. In response to heat stress, ERK1/2 initiates a series of cascading reactions that modulate the balance between cellular survival and apoptosis molecules, thereby safeguarding a portion of male germ cells and somatic cells within the testis from destruction. Research indicates that a brief 30 min exposure to heat stress triggers an increase in phosphorylated ERK1/2 (pERK1/2) levels in immature boar Sertoli cells. This, in turn, elevates HSP70 levels and subsequently enhances the production of lactate, a primary ATP substrate crucial for the development of germ cells, by accelerating glucose metabolism [
127]. Furthermore, ERK signaling exerts protective effects on pachytene spermatocytes subjected to transient heat stress by upregulating metastasis-associated 1, which counteracts the pro-apoptotic effects of p53 [
128]. Conversely, inhibiting the ERK1/2 signaling pathway during heat stress significantly reduces the expression of genes such as c-fos, AP-1, and ERK2, as well as the phosphorylation of ERK1/2 and c-Fos. This inhibition is accompanied by a marked increase in c-Jun mRNA expression within Sertoli cells. Notably, the adverse effects of heat stress on the ERK1/2 signaling pathway can be ameliorated through treatment with baicalin [
113]. Consistently, Wang et al. have reported that heme oxygenase 1 (HO-1) utilizes the ERK1/2 signaling pathway to suppress the expression of apoptotic genes, specifically Bax/Bcl-2, thereby restoring the normal functionality of bovine granulosa cells [
111]. This interplay between ERK1/2 signaling and HO-1 underscores their pivotal roles in modulating cellular responses to heat stress, ultimately influencing cell survival and apoptosis in reproductive cells.
3.4. Protective Role of Nrf2 in Protection of Mammalian Cells against Heat-Stress-Induced Oxidative Stress and Apoptosis
Nrf2 is an inducible transcription factor crucial for maintaining redox signaling integrity in the face of oxidative stress [
107]. Nrf2, a member of the Cap’n’Collar basic leucine zipper transcription factor family, plays a pivotal role in orchestrating antioxidant and detoxification responses through the upregulation of its downstream genes [
129]. In unstressed cells, the Nrf2 is primarily located within the cellular cytoplasm and forms a complex with its inhibitory partner, Kelch-like ECH-associated protein 1 (Keap1). However, when the cellular environment encounters an elevated presence of ROS, the Keap1-Nrf2 complex undergoes dissociation, leading to the translocation of Nrf2 from the cytoplasm into the cellular nucleus [
130,
131]. Furthermore, the activated Nrf2 binds to the antioxidant response element (ARE) sequence, thereby stimulating the transcription of genes involved in antioxidant defenses and neutralizing ROS-induced damage [
115]. Recent findings indicate that p62 can competitively interact with Keap1 at the Nrf2 binding site, altering the association, releasing ubiquitinated Nrf2, and ultimately activating the Nrf2 antioxidant systems [
132,
133,
134].
Heat-stress-induced apoptosis triggers antioxidant responses, including autophagy and Nrf2 activation [
135]. It has been observed that changes in autophagy dynamics are pivotal regulators of the Nrf2 signaling pathway’s protective role in the testis. This protection is achieved by suppressing MDA levels and promoting an antioxidant status that shields the testis from the adverse consequences of heat stress [
136,
137,
138,
139]. Notably, inhibiting Nrf2 in cells results in reduced cell viability, elevated MDA levels, and Sertoli cell death [
107].
Nrf2 is known to regulate several crucial antioxidant genes, including catalase (CAT), heme oxygenase 1 (HMOX1), peroxiredoxin 1 (PRDX1), SOD1, and thioredoxin 1 (TXN1). These genes collectively enhance antioxidant activity, mitigating oxidative stress in mouse testis cells and protecting germ cells and Leydig cells from oxidative damage [
56,
139]. Furthermore, recent research has shown that heat-stress-induced ROS overproduction suppresses the expression of antioxidant genes (SOD, CAT, NQO1, and GSH-Px) in uterine tissue [
98]. In Sertoli cells, elevated ROS levels due to heat stress increase MDA levels and decrease antioxidant enzyme levels [
140]. Additionally, heat stress has been found to increase the expression of apoptotic markers such as Fas, FasL, caspase 3, and caspase 9 in mouse Sertoli cells [
140]. Consequently, the Keap1/Nrf2 signaling pathway has been significantly associated with the protective effects observed in mouse uterine tissue, marked by increased levels of antioxidant genes [
98].
Moreover, oxidative stress influences various important signaling pathways, including the nuclear factor erythroid 2-related factor 2 (Nrf2)/Keap1 signaling axis in the testis [
141]. A recent study highlights Nrf2’s protective role in safeguarding mouse Sertoli cells from heat-induced oxidative stress through the Nrf2/Keap1 signaling pathway [
107]. Similarly, another investigation revealed that Nrf2 significantly reduces caspase 3 levels, subsequently reducing cell death induced by heat stress treatment in Sertoli cells [
137]. Under conditions of severe heat stress, the heightened expression of Keap1 and NFE2L2 facilitates the regulation of genes associated with antioxidants by forming complexes with the ARE, thus establishing a defensive mechanism against heat stress within bovine endometrial epithelial cells [
142]. These findings collectively underscore the critical role of Nrf2 in mitigating oxidative stress and apoptosis in various cellular contexts, particularly under heat stress conditions.
3.5. Role of Adenosine 5′-Monophosphate-Activated Protein Kinase (AMPK) in Self-Recovery from Heat-Stress-Induced Oxidative Stress and Apoptosis
It is well established that AMPK signaling plays a key role in the tight junctions (TJs) and cell proliferation of testis Sertoli cells [
143,
144,
145,
146,
147,
148]. In addition, Ni et al. [
147] highlighted that Sertoli cells play a key role in lactate supply, maintenance of cell junctions, and support for germ cells’ mitosis and meiosis. The AMPK signaling pathway regulates the dynamics of tight junctions and adherens junctions; the proliferation and meiosis of germ cells; and the energy metabolism, proliferation, and lactate production of Sertoli cells [
149]. Once this balance is disrupted, the microenvironment of the testis and the quality of sperm will be affected. When α1AMPK was conditionally knocked out in mouse SCs, the mutant mice still showed an abnormal phenotype, including thin-head spermatozoa, reduced expression of junctional proteins (β-catenin, vimentin, occludin, and ZO-1), and deregulation of energy homeostasis [
143,
145,
147,
148]. Consistently, a study has documented that curcumin (natural antioxidant and anti-inflammatory compound) supplementation rescues porcine Sertoli cell impairment and TJs by inhibiting the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome through the AMPK/SIRT3/SOD2/mtROS signaling pathway [
150]. Heat stress can cause dysfunction of TJs in porcine testis reversibly via Ca
2+/calmodulin-dependent protein kinase kinase B (CaMKKB)-induced inhibition of the AMPK signaling pathway. Consistently, Yang et al. [
148] treated SCs from 3-week-old piglets at 43 °C for 0.5 h, and this hyperthermia treatment inhibited the AMPK signaling pathway, inhibiting the expression of CLDN11, JAMA, occludin, and especially ZO-1 in porcine SCs [
148]. In addition, it was observed that normal Sertoli cell function was restored after 48 h due to AMP signaling [
145].
This entry is adapted from the peer-reviewed paper 10.3390/antiox13030258