Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Human epidermal growth factor receptor 2 (HER2) tyrosine kinase is overexpressed in 20% of breast cancers and associated with a less favorable prognosis compared to HER2-negative disease.
- HER2
- antibody–drug conjugates
- breast neoplasms
- targeted therapy
1. Introduction
Breast cancer is the most common malignancy among women and a leading cause of cancer-related deaths worldwide [1]. There are three main molecular subtypes of breast cancer, which differ significantly in natural history, prognosis, and treatment options. These are commonly known as hormone receptor-positive/HER2-negative (luminal A, luminal B), human epidermal growth factor receptor 2 (HER2)-positive, and triple-negative [2]. The HER2 protein is overexpressed in 20–30% of breast cancers and is historically associated with higher cancer recurrence rates and shorter disease-free and overall survival when compared with HER2-negative breast cancers [3]. HER2 is a member of the epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases, which includes EGFR/HER1, HER2, HER3, and HER4. These receptors play a significant role in cell growth and differentiation. Overexpression of HER2 is linked to upregulated cell proliferation and the development of several cancers, including breast, gastric, and ovarian cancer [3,4]. Identification of the HER2 status is achieved by two methods: immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH). All patients with invasive breast cancer have the tumor evaluated first by IHC (graded from 0 to 3+), followed by validation with FISH for borderline (IHC 2+) results. Patients with tumors that are IHC 3+- or FISH-positive tend to respond to anti-HER2 therapies. HER2 testing methodologies have evolved over time, including different ways of assessing HER2 protein expression using immunohistochemistry with novel scoring systems (e.g., H-score). HER2-Low breast cancer represents a new subset of breast cancers that may respond to HER2 ADC therapy. The development and validation of quantitative assays to determine HER2 protein levels in breast cancer tissue samples is an area of active investigation [5].
The approval of trastuzumab, a monoclonal antibody targeting HER2, in 1998 marked a significant advancement in the therapeutic landscape for HER2-positive breast cancer [6]. Subsequently, there has been a substantial increase in the clinical development of innovative therapies aimed at this specific breast cancer subtype [7]. The strategic incorporation of monoclonal antibodies and tyrosine kinase inhibitors, along with chemotherapy, endocrine therapy, and immunotherapy, has profoundly transformed the clinical outcomes for individuals diagnosed with HER2-positive breast cancer [8]. This paradigm shift has occurred at both the early and advanced stages of the disease [9].
While monoclonal antibodies and tyrosine kinase inhibitors represent major advances in the treatment of HER2-positive breast cancer, a significant number of patients with early breast cancer develop metastatic disease despite adjuvant systemic therapy. Furthermore, the development of progressive disease in the metastatic setting and the lack of curative therapies remain unmet needs. Understanding molecular mechanisms of resistance in HER2-positive breast cancer cells, together with improvements in technology, led to the development of antibody–drug conjugates (ADCs). These drugs represent an advanced modality in oncological treatment, integrating the specificity of monoclonal antibodies with the cytotoxic power of potent drugs, also known as “payloads”, using specific linkers [10].
The chemical structure of ADCs consists of three integral elements: a monoclonal antibody, a chemical linker with stability or cleavability properties, and a cytotoxic payload/agent (Figure 1).
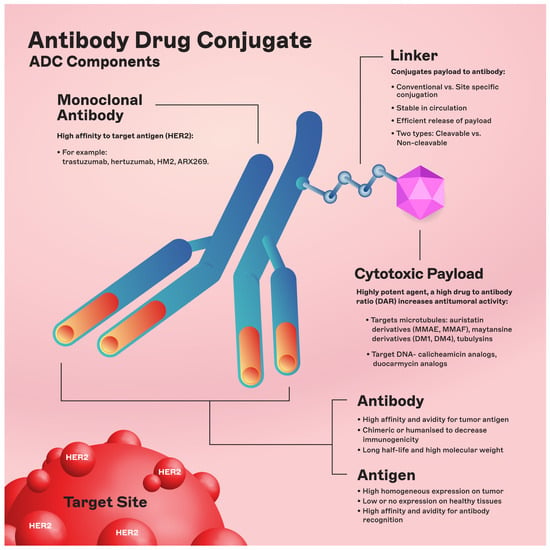
Figure 1. ADC components. Abbreviations: DAR: drug-to-antibody ratio; DM1: derivative of maytansine 1; DM4: derivative of maytansine 4; MMAF: monomethyl auristatin F; MMAE: monomethyl auristatin E.
The engineered monoclonal antibody (e.g., trastuzumab) is designed to detect and bind to an antigen expressed on the surface of cancer cells (i.e., HER2) [11]. Following attachment, the antibody–drug complex is internalized via receptor-mediated endocytosis [12]. While pinocytosis may also facilitate ADC uptake in the absence of the target antigen, the conjugated antibody’s considerable size and hydrophilic character significantly mitigate nonspecific absorption, thereby augmenting the specificity and safety of ADCs [13]. Upon cellular entry, the ADC is trafficked to endosomes and lysosomes, where enzymatic cleavage of the linker ensues, resulting in the release of the cytotoxic payload. This release enables the drug to unleash its cell-killing potential (Figure 2). The therapeutic agents employed in ADCs are diverse, ranging from microtubule disruptors to topoisomerase inhibitors. A pivotal feature of ADCs is the “bystander effect,” wherein the liberated toxins can permeate and exterminate neighboring tumor cells that may not express the target antigen, thereby increasing the antitumor effect [14].
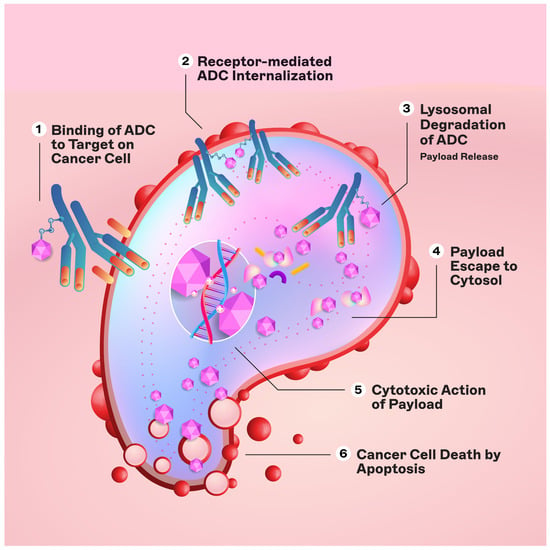
Figure 2. Mechanism of action of ADCs.
2. Current Landscape of HER2-Targeted ADCs
To date, two ADCs directed against HER2—trastuzumab emtansine (T-DM1, Kadcyla) [15] and trastuzumab deruxtecan (T-DXd, DS-8201a, Enhertu) [16]—have been approved by the Food and Drug Administration (FDA) and other regulatory agencies throughout the world for the management of HER2-positive breast cancer. In addition, T-DXd was approved for patients with HER2-Low metastatic breast cancer (IHC score 1 or IHC 2 with negative FISH) (Table 1) [17]. The DESTINY-Breast06 trial is testing the efficacy of T-DXd in patients with HER2-Ultra-Low breast cancer (IHC score 0, with 1–10% cells staining weakly).
Table 1. Key characteristics of selected HER2 ADCs.
| ADC Name | mAb | Payload | Linker | DAR | Clinical Phase | References |
|---|---|---|---|---|---|---|
| Trastuzumab emtansine (T-DM1) |
Trastuzumab | DM1 | Non-cleavable SMCC linker | 3.5 | Approved for metastatic HER2-positive breast cancer, residual disease after neoadjuvant therapy | [18,19] |
| Trastuzumab deruxtecan (DS-8201a) | Trastuzumab | DXd | Cleavable GGFG linker | 8 | Metastatic HER2-positive and HER2-Low breast cancer | [16,20] |
| Trastuzumab duocarmazine (SYD985) | Trastuzumab | seco-DUBA | Cleavable vc linker | 2.7 | Phase I/II—advanced breast cancer | [21] |
| ARX-788 | Anti-HER2 mAb (ARX269) | MMAF | Non-cleavable linker conjugated to pAcF | 1.9 | Phase II—advanced breast cancer | [22] |
| ALT-P7 | Trastuzumab biobetter (HM2) | MMAE | Cleavable cysteine-containing peptide | 2 | Phase I | [23] |
| BL-M07D1 | Trastuzumab | Ed-04 | Cathepsin B cleavable linker | 8 | Phase I—advanced breast cancer | [24] |
| Disitamab vedotin (RC48) | Hertuzumab | MMAE | mc-val-cit PABC linker | 4 | Phase I | [25] |
| Zanidatamab zovodotin | Zanidatamab | Zovodotin | Cleavable vc linker | 2–4 (variable) | Phase II | [26] |
Abbreviations: mAb: monoclonal antibody; DAR: drug-to-antibody ratio; DM1: derivative of maytansine 1; SMCC: succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate; DXd: derivative of exatecan; GGFG: glycine–glycine–phenylalanine–glycine; vc: valine–citrulline; seco-DUBA: seco-duocarmycin hydroxybenzamide azaindole; MMAF: monomethyl auristatin F; pAcF: para-acetylphenylalanine; MMAE: monomethyl auristatin E; mc-val-cit PABC: maleimidocaproyl–valine–citrulline–p-aminobenzylcarbamate.
2.1. Trastuzumab Emtansine (T-DM1)
Trastuzumab emtansine (T-DM1) represents a seminal advancement in the treatment of HER2-positive breast cancer. T-DM1 (Kadcyla) was the first ADC to receive FDA approval in the U.S. in 2013 for single-agent treatment for advanced HER2-positive breast cancer (following treatment with trastuzumab and a taxane). Its approval was expanded in 2019 for the use of T-DM1 for patients with early-stage high-risk HER2-positive breast cancer with residual disease after neoadjuvant therapy with trastuzumab and taxane-based treatment [19].
The T-DM1 molecule consists of a cytotoxic component (Emtansine) attached to the antibody trastuzumab through a stable linker. Maytansine is a highly potent cytotoxic agent derived from the Ethiopian plant Maytenus serrata. Due to its high toxicity, it is not used directly as a cancer treatment but rather as a part of a targeted therapy. Emtansine (also known as DM1) is a derivative of maytansine that has been chemically modified to be less toxic and more stable in the bloodstream. T-DM1 retains trastuzumab’s inhibitory functions—especially the blockade of the PI3K/AKT pathway. In addition, trastuzumab facilitates T-DM1′s internalization and subsequent disintegration to unleash the potent microtubule-inhibitory action of the MCC-DM1 complex [27].
The EMILIA trial showcased the superiority of T-DM1, demonstrating its ability to improve progression-free survival (PFS) rates compared to a regimen that combined lapatinib and capecitabine in patients with HER2-positive metastatic breast cancer who had received a prior taxane [15]. In this trial, median progression-free survival was improved from 6.4 months with lapatinib–capecitabine to 9.6 months with T-DM1. Overall survival and objective response rates were also improved with T-DM1. The KATHERINE trial showed improvement in disease-free survival (DFS) rates in patients with early-stage HER2-positive breast cancer with residual disease following neoadjuvant trastuzumab-based treatment [19]. At a follow-up of 3 years, the percentage of patients who were free of invasive disease was 88.3% in the T-DM1 group compared with 77.0% in the trastuzumab group. This randomized trial validated the role of T-DM1 for patients with residual disease after neoadjuvant therapy when compared to continued trastuzumab therapy.
Furthermore, in the KRISTINE trial, T-DM1 therapy was compared to sequential anthracycline-based chemotherapy followed by taxane in combination with trastuzumab and pertuzumab, or the TCHP (docetaxel, carboplatin, trastuzumab, pertuzumab) regimen in the neoadjuvant setting. In this study, T-DM1 demonstrated a reduced pathologic complete response rate compared to the other regimens [28]. Nevertheless, the KRISTINE trial linked it to a more favorable safety profile, achieving pathologic complete responses in 44% of patients without conventional chemotherapy.
2.1.1. Mechanisms of Resistance to T-DM1
The mechanisms of resistance to T-DM1 in breast cancer are multifactorial and occur via various different pathways.
-
Antigen-Related Resistance Mechanisms. T-DM1 resistance can develop via various pathways, particularly in JIMT1 cells, which are inherently resistant to first-line trastuzumab [29]. These cells, upon TM-ADC treatment, showed resistance while remaining sensitive to other chemotherapeutics. It suggests that prolonged exposure to HER2-targeted therapy could decrease HER2 levels, leading to treatment-resistant cells [30]. Additionally, heterogeneity in HER2 expression, as observed in the KRISTINE and ZEPHIR trials, correlated with lower efficacy of T-DM1, marked by no pathologic complete responses and shorter progression-free survival (PFS) and overall survival (OS) [31,32]. Truncated forms of the antigen ectodomain, like P95HER2, and antigen masking by molecules such as MUC4 have also been implicated in resistance [33]. Furthermore, ligand-induced heterodimerization of HER2 with other receptors can impair T-DM1’s effectiveness [29].
-
Payload-Related Resistance. Tumor cells may develop resistance to the cytotoxic drug (DM1). T-DM1-resistant cells with upregulated ABC transporter expressions (ABCC2, ABCG2) exhibited reduced sensitivity, which could be countered by inhibiting these transporters [34]. The diversity in payloads, conjugation sites, and drug-to-antibody ratios (DAR) also significantly impacts ADC efficacy, suggesting that ADC resistance can be payload specific [35,36].
-
Internalization and Trafficking Pathways. T-DM1 is internalized into cancer cells via endocytosis. Variations in endocytic routes, regulated by specific proteins, can affect ADC delivery and processing. For instance, some T-DM1-resistant cells have been shown to internalize ADCs into caveolin-1-coated vesicles, indicating an alternative trafficking pathway. Proteins like Endophilin A2 also play a role in HER2 internalization, affecting T-DM1 sensitivity [37].
-
Lysosomal Dysfunction. After T-DM1 internalization, lysosomal cleavage releases the cytotoxic drug. Any disruption in lysosomal function, such as altered pH or proteolytic activity, can hinder this process. Resistant clones with higher lysosomal pH and accumulated T-DM1 have been documented, indicating impaired ADC processing [38]. The transport of cytotoxic drugs from lysosomes to the cytoplasm, especially relevant for non-cleavable linkers, is another potential resistance mechanism [39]. In the DAISY trial, a phase II multicenter, open-label study, researchers investigated resistance to T-DXd in three distinct patient groups: HER2-positive, HER2-zero, and low-HER2. Participants received T-DXd at 5.4 mg/kg triweekly, aiming for the best objective response rate as the primary measure of success. When the cancer progressed, whole-genome sequencing was employed to uncover potential resistance mechanisms. The findings indicated that apart from reduced HER2 expression, alterations in the SLX4 gene could contribute to resistance. SLX4 is integral to DNA damage repair, overseeing three endonucleases. The evidence showed that a deficit in SLX4 correlated with resistance to T-DXd, suggesting that loss-of-function mutations in SLX4 could be implicated in the development of resistance to the drug [40].
-
Drug-Efflux Mechanisms. Overexpression of ABC transporters, which increase drug efflux from cells, can contribute to resistance. For example, maytansinoids, T-DM1’s payload, are known substrates of ABC transporters like MDR1, linking resistance to increased expression of these transporters [41].
-
Cell Cycle Dependencies. The cell cycle status affects T-DM1 effectiveness. Resistance to T-DM1 has been linked to variations in cyclin B levels, a cell cycle regulator. Accumulation of cyclin B1 in sensitive cells, as opposed to resistant ones, suggests that cell cycle dysregulation can modulate T-DM1 efficacy [42].
-
Activation of Survival Signaling Pathways. Activation of pathways like PI3K/AKT/mTOR, which are involved in cell survival, can decrease sensitivity to trastuzumab-based therapy. PTEN loss or PIK3CA hyperactivation can lead to reduced trastuzumab sensitivity [43]. However, T-DM1 may remain effective even with these mutations, as indicated by the EMILIA trial results [44]. Therefore, this is an area of ongoing investigation.
-
Apoptosis Dysregulation. Finally, changes in apoptosis regulation, such as the overexpression of proteins like BCL-2 and BCL-XL, have been correlated with resistance to ADCs like gemtuzumab ozogamicin [45] and brentuximab vedotin [46], indicating a potential mechanism of resistance to other ADCs like T-DM1, although this mechanism of resistance is not well defined in breast cancer.
2.1.2. T-DM1 Combination Therapies
Current research strives to devise approaches to surmount these resistance mechanisms to improve patient prognosis. Personalized medicine strategies, including targeted and immune-based therapies, are under exploration to effectively counter resistance. In efforts to surmount resistance to T-DM1 in treating HER2-positive breast cancer, the integration of T-DM1 with diverse therapeutic agents has been extensively investigated. This strategy targets specific resistance mechanisms, proposing alternate modalities to boost treatment efficacy.
Many agents have been assessed for their potential synergistic effects with T-DM1, including monoclonal antibodies (e.g., pertuzumab), tyrosine kinase inhibitors (e.g., lapatinib, neratinib, tucatinib), PI3K pathway inhibitors (e.g., alpelisib), PD1/PDL1 checkpoint inhibitors (e.g., pembrolizumab, atezolizumab) as well as CDK4/6 inhibitors (e.g., palbociclib, ribociclib, abemaciclib) [47,48,49,50,51].
The logic behind these combinations is to mount a diversified onslaught on HER2-positive breast cancer cells, targeting various pathways and resistance mechanisms. Utilizing T-DM1 with these agents is anticipated to improve treatment results and confront the hurdles posed by drug resistance. Future clinical studies will shed light on the success of these combination treatments for patients with HER2-positive breast cancer.
2.1.3. T-DM1 Toxicities and Safety Profile
With the development of new ADCs comes specific toxicities. In the landmark EMILIA trial [15], 15.5% of patients experienced a grade 3 adverse event (compared with 18% in the lapatinib–capecitabine arm). The most common grade 3 and 4 events with T-DM1 were thrombocytopenia (12.9%) and liver enzyme elevations of aspartate aminotransferase (4.3%) and alanine aminotransferase (2.9%). In the EMILIA trial, the occurrence of grade 3 or 4 thrombocytopenia was most common during the first two cycles of T-DM1 treatment, and the majority of patients were able to continue treatment with dose modifications. Overall, the incidence of bleeding was more common among patients treated with T-DM1 (29.8%) compared with lapatinib–capecitabine (15.8%), but rates of grade 3 or 4 bleeding were low in both groups (1.4% and 0.8%, respectively). One grade 4 gastrointestinal bleed did occur in a patient on T-DM1 but whose platelet count was within the normal range at that time. Additionally, most patients were able to continue treatment with dose modifications for elevated liver enzymes as well. These side effects highlight the importance of vigilant laboratory monitoring throughout the therapy [15,19,28].
Cardiotoxicity was also evaluated in the EMILIA trial, and the majority of patients treated with T-DM1 maintained an ejection fraction greater than or equal to 45% while on therapy (97.1%) [15]. Only one patient treated at the time of publication had developed grade 3 left ventricular systolic dysfunction after treatment with T-DM1. Baseline ejection fraction should be assessed and monitored throughout treatment while on T-DM1.
2.2. Trastuzumab Deruxtecan (T-DXd)
Trastuzumab deruxtecan (T-DXd) is a novel ADC that has transformed the treatment of breast cancer. T-DXd distinguishes itself by utilizing a topoisomerase I inhibitor derivative (i.e., deruxtecan) as its payload, connected via a cleavable tetrapeptide-based linker [20]. This cleavable linker is selectively severed within tumor cells, reducing off-target release and associated toxicities. T-DXd possesses a notably higher drug-to-antibody ratio (DAR) relative to T-DM1, an attribute that contributes to its potent efficacy [52].
Clinical investigations, including the pivotal DESTINY-Breast01 and DESTINY-Breast03 trials, have demonstrated T-DXd’s efficacy in significantly prolonging progression-free survival (PFS) over T-DM1, leading to its endorsement by the FDA as a second-line therapy for HER2-positive metastatic breast cancer [16,53]. Furthermore, T-DXd’s approval for HER2-Low breast cancer signals the recognition of a new subset within the breast cancer spectrum, expanding the therapeutic landscape [54].
While T-DXd has shown promising clinical efficacy in HER2-positive breast cancer, its use is accompanied by potential adverse effects that require vigilant monitoring. Paramount among these is the risk of drug-related interstitial lung disease (ILD) or pneumonitis, inflammatory conditions that may progress to severe impairment of lung function and significant respiratory compromise. Other toxicities include gastrointestinal symptoms, hematological abnormalities, and rare cardiac toxicities. Regular monitoring and appropriate supportive treatments are essential for managing these side effects [16].
2.2.1. T-DXd: Mechanisms of Resistance
Trastuzumab deruxtecan (T-DXd) has emerged as a significant therapeutic agent in the treatment of HER2-positive and HER2-Low metastatic breast cancer. Nonetheless, resistance to T-DXd poses a significant obstacle in clinical settings. A thorough understanding of the resistance mechanisms is crucial to improving therapeutic strategies and subsequent patient outcomes.
-
HER2 Receptor Modifications: Modifications of the HER2 receptor are a primary resistance mechanism, including mutations, gene amplification, or structural alterations, which can diminish the receptor’s affinity for T-DXd. These modifications may reduce the drug’s efficacy by impairing target binding, necessitating the investigation of methods to overcome these changes in HER2 [55].
-
ADC Internalization and Intracellular Trafficking: The internalization and intracellular trafficking of T-DXd are critical to its cytotoxic action. Resistance may develop from disruptions in these processes, impeding the delivery of the cytotoxic payload. Enhancing T-DXd internalization and trafficking could be a strategic approach to bypass this resistance mechanism [56].
-
Drug-Efflux Transporters: The expression of efflux transporters, such as P-glycoprotein (P-gp), which expel the payload (deruxtecan) from cells, can decrease its intracellular concentration and cytotoxic impact. Inhibition or circumvention of these transporters is under investigation to enhance deruxtecan’s intracellular retention [34].
-
Tumor Microenvironment and Stromal Factors: The tumor microenvironment, including stromal cell-secreted factors, can confer survival advantages to cancer cells, fostering resistance to T-DXd. Targeting the tumor microenvironment through combination therapies or immune-modulating agents may address this resistance mechanism [57]. In the DAISY trial evaluating the efficacy of T-DXd in breast cancer patients at different levels of HER2 expression, researchers evaluated the impact of T-DXd on the tumor microenvironment. This exploratory study included samples collected from 31 patients. No significant changes in immune cell levels were observed at the 3-week or 6-week mark following treatment. However, a notable reduction in PD-L1 expression among the first cohort of patients was attributed to T-DXd’s cytotoxic effects on PD-L1-positive tumor cells. Additionally, a decline in the presence of macrophages close to tumor cells was reported in the same cohort [40].
-
Alternative Signaling Pathways: The activation of alternative signaling pathways, such as the PI3K/AKT/mTOR pathway, can provide survival advantages to cancer cells, undermining T-DXd’s effectiveness. Co-targeting HER2 and these alternative pathways may be essential to counteract resistance [7,43,58,59].
-
Tumor Heterogeneity: The intrinsic heterogeneity of tumors, both among patients and within a single tumor, can result in cancer cell populations with varying sensitivities to T-DXd, contributing to the complex nature of resistance. Personalized treatments, considering the unique molecular and phenotypic profiles of tumors, may be promising in overcoming resistance [60].
In summary, resistance to T-DXd in breast cancer is complex and involves diverse interactions between the tumor cells and their surrounding microenvironment. Current research is focused on elucidating these mechanisms in detail and developing targeted strategies to counter resistance. Anticipated advancements in T-DXd-based treatments for HER2-positive and HER2-Low breast cancer include personalized medicine, combination therapies, and an enhanced understanding of tumor biology.
2.2.2. T-Dxd Combination Therapies
Therapeutic combinations are being rigorously evaluated to surmount resistance to T-DXd in breast cancer therapy. These research efforts aim to refine treatment regimens and enhance patient prognoses. A range of combinations are in various stages of clinical trials, following the same paradigm as T-DM1 above. The objectives of these clinical trials are to establish the safety profiles, efficacy, and appropriate dosing regimens for these combination treatments. The forthcoming results are expected to yield critical insights into the most efficacious combinations and patient demographics best suited for these therapies. The overarching aim is to personalize treatment approaches, thereby advancing the care and outcomes for patients with HER2-positive breast cancer who exhibit resistance to T-DXd.
2.2.3. T-DXd in HER2-Low Breast Cancer
In the realm of breast cancer treatment, the categorization of tumors as HER2-positive or HER2-negative has been traditionally binary. However, a subset of tumors exhibit low levels of HER2 (“HER2-Low”), which is found in 45–60% of cases without HER2 amplification or overexpression [5]. These HER2-Low tumors are identified by an immunohistochemical (IHC) score of 1+ or a score of 2+ accompanied by a negative in situ hybridization (ISH) result. In the pivotal DESTINY-Breast 04 trial, trastuzumab deruxtecan (T-DXd) showed significant effectiveness in treating HER2-Low metastatic breast cancer, achieving a 52.6% objective response rate among patients who had undergone one or two prior lines of therapy [17].
While HER2-0 breast cancers are often less amenable to monoclonal antibody therapy, a subset known as HER2-Ultra-Low has been recognized, characterized by minimal HER2 protein expression. Ongoing studies are exploring the use of ADCs for this group. For example, the DESTINY-Breast06 trial is investigating the efficacy of T-DXd in patients with HER2-Ultra-Low metastatic breast cancer. Additionally, certain genetic mutations, like the V777L ERBB2 mutation and MutL deficiency—related to mismatch repair system changes—suggest potential responsiveness to anti-HER2 therapies, even in HER2-negative breast cancers.
Ongoing research is assessing T-DXd versus chemotherapy in hormone receptor-positive, HER2-Low metastatic breast cancer and exploring its combination with immune checkpoint inhibitors. Early data indicate favorable safety and efficacy, with high response rates [61]. The combination of T-DXd with immune therapies such as PD-L1 and PD-1 inhibitors is being evaluated, showing promising activity, although questions about the incremental benefit over T-DXd alone persist. These studies underscore the potential of T-DXd as a key therapeutic for HER2-Low breast cancer, offering hope for improved outcomes in this diverse patient population.
2.2.4. T-Dxd Toxicities and Safety Profile
In the DESTINY-Breast03 trial, 45.1% of patients receiving T-DXd experienced any grade 3 or 4 adverse effects. The most common drug-related toxicities in the T-DXd arm were nausea (72.8%), fatigue (44.7%), and vomiting (44%). Overall, T-DM1 was better tolerated. Drug-related alopecia was also common, occurring in 36.2% of patients who received T-DXd (compared with 2.3% in patients treated with T-DM1). Neutropenia (19.1%), thrombocytopenia (7%), and leukopenia (6.6%) were also noted. Cardiotoxicity, as with T-DM1, appeared to be a rare event, but echocardiograms should be monitored.
The most concerning of the adverse events associated with T-DXd remains interstitial lung disease (ILD), or pneumonitis, which occurred in 10.5% of patients treated with T-DXd. The median time to onset of ILD/pneumonitis was 168 days (range: 33 to 507). Treatment was discontinued in 8.2% of patients treated with T-DXd due to pneumonitis. Fatal cases of ILD had occurred in earlier trials; however, most patients experience grade 1 or 2 events. Increased awareness has led to improved monitoring and treatment of this rare but serious side effect. Risk factors for the development of pneumonitis/ILD include dose >6.4 mg/kg, age <65, baseline oxygen saturation <95%, moderate to severe kidney impairment, pulmonary comorbidities (asthma, COPD, prior ILD/pneumonitis, pulmonary fibrosis, emphysema, and radiation pneumonitis) and >4 years since initial diagnosis [62].
Experts recommend high-resolution chest CT every 6 months for monitoring for pneumonitis and ILD in patients undergoing treatment with T-DXd, if available. If ILD is suspected, the drug must be held, and steroids should be promptly administered, in addition to pulmonary consultation and evaluation. If grade 1 toxicity occurs, the patient can be rechallenged with the drug. If grade 2 (symptomatic) toxicity occurs, the drug must be held permanently.
This entry is adapted from the peer-reviewed paper 10.3390/cancers16040800
This entry is offline, you can click here to edit this entry!