Involvement of the immune system in biological therapies specifically targeting tumor microenvironment has been suggested. Substantial advancement in the treatment of malignant tumors utilizing immune cells, most importantly T cells that play a key role in cell-mediated immunity, have led to success in clinical trials.
- tumor microenvironment
- cancer vaccines
- tumor-infiltrating lymphocytes
- combination therapy
- checkpoint-inhibitors
- CAR T cell therapy
1. Tumor Microenvironment in Cancer
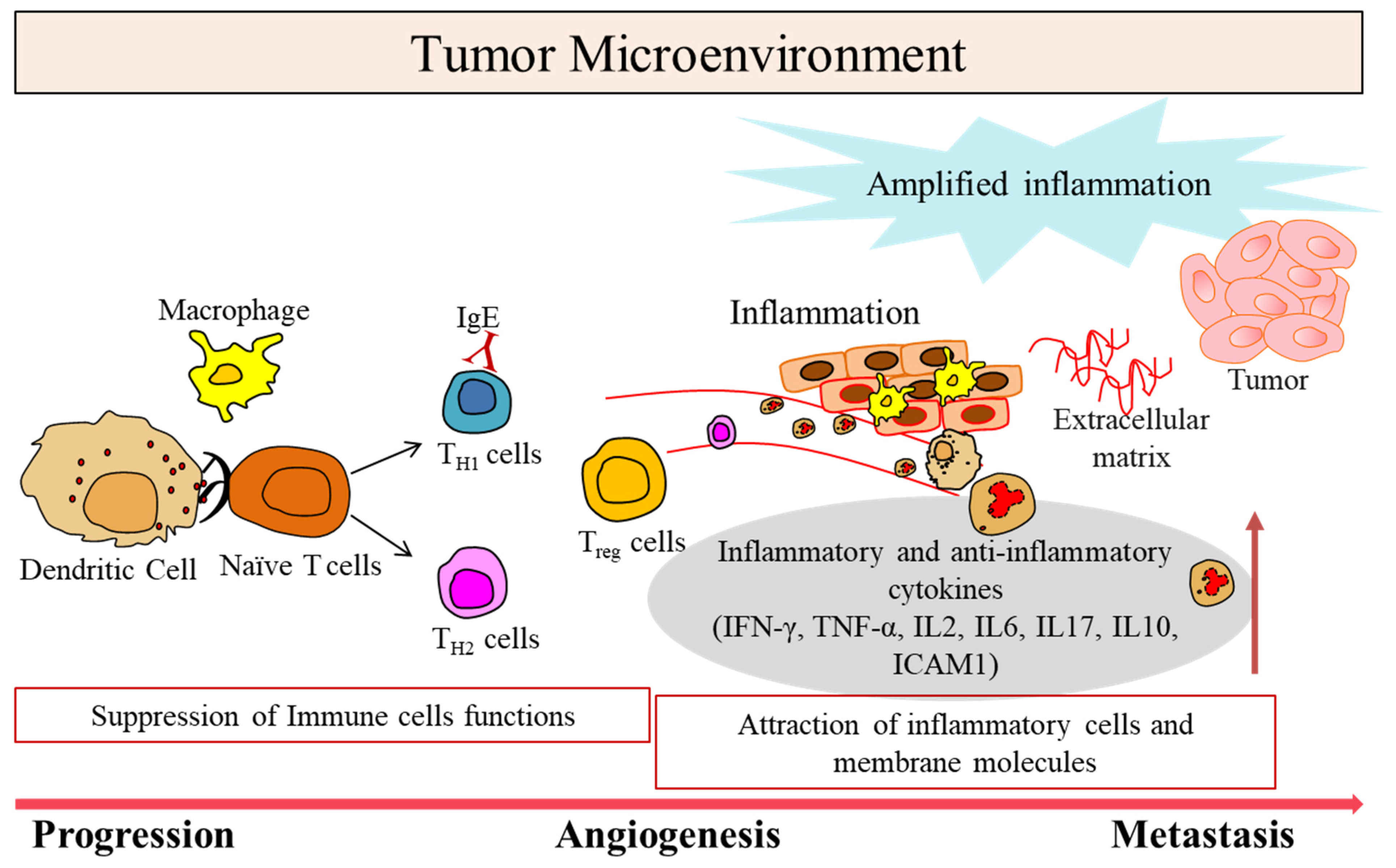
2. Immunomodulatory Roles of Lymphangiogenesis in TME
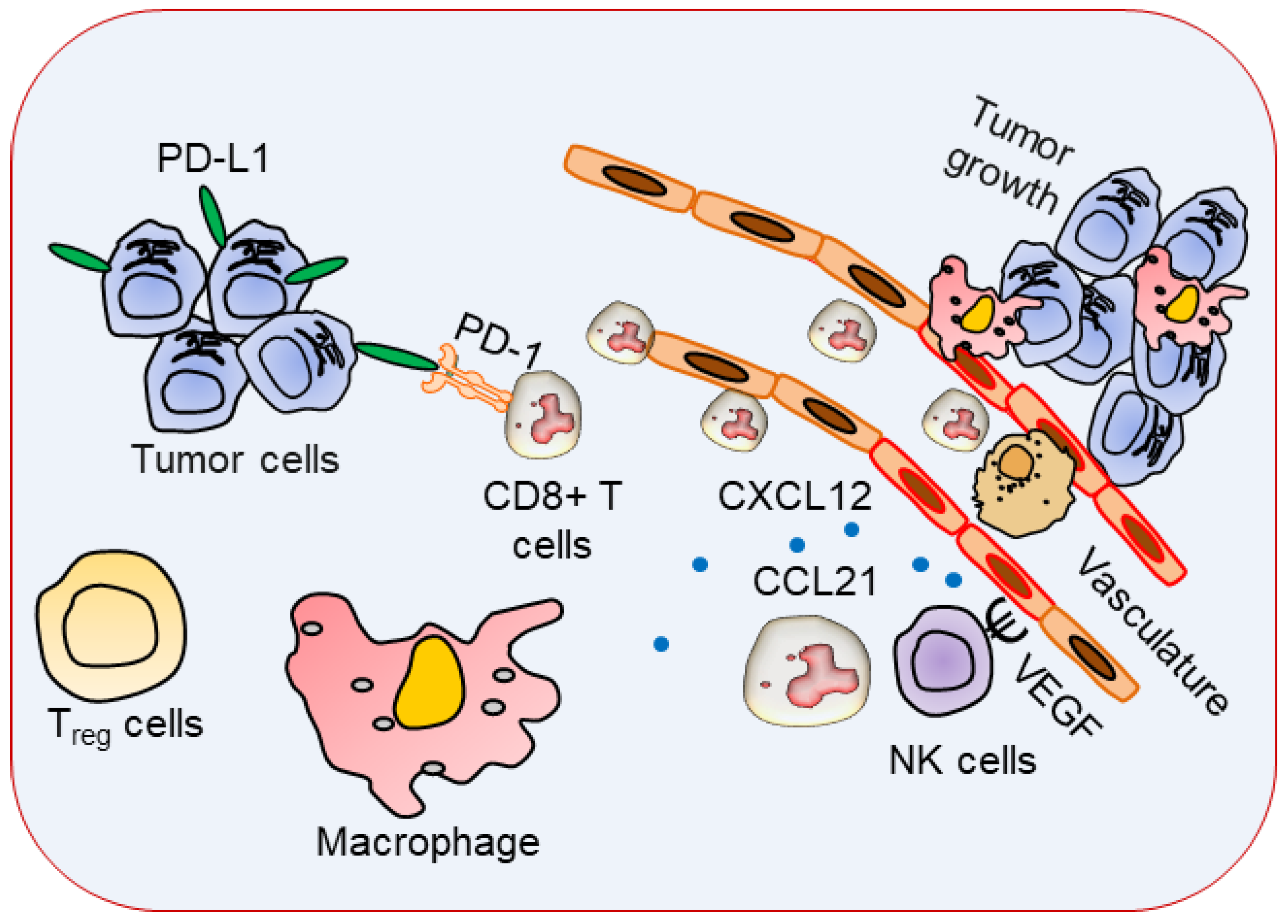
3. Immune Cells with Specific Phenotypes in TME
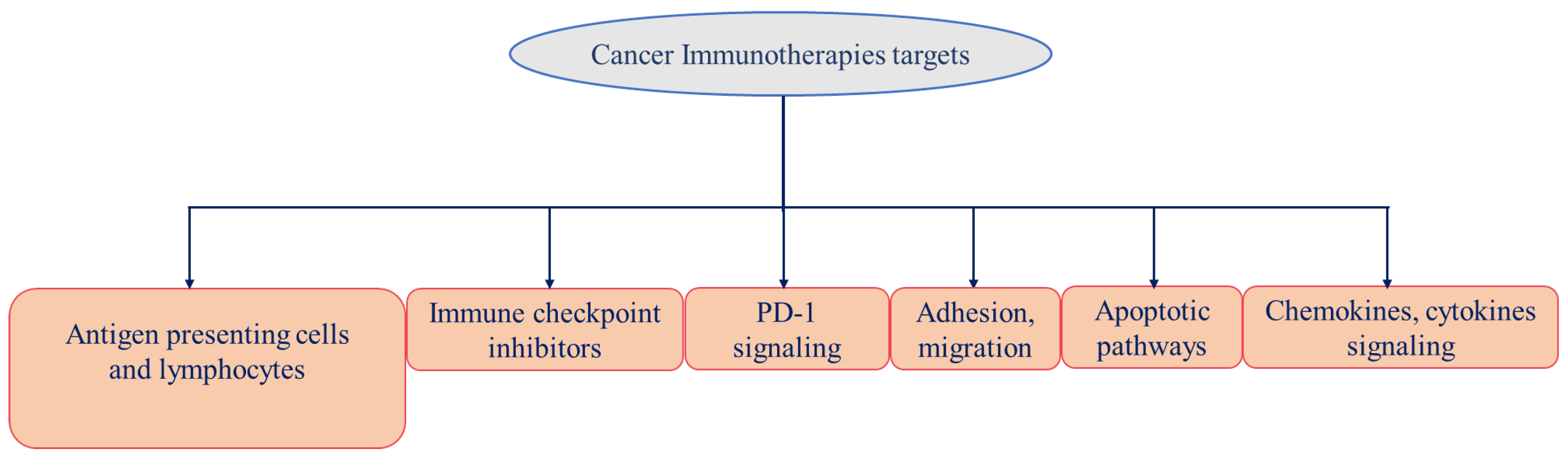
4. Cancer Immunotherapy
|
Target |
Drug INN (Brand Name) |
Cancer Type |
Current Status |
Ref. |
|---|---|---|---|---|
|
Antibodies Based |
||||
|
CTLA-4 |
Ipilimumab (Yervoy) |
Melanoma (2011) and Renal cell carcinoma (2018) |
FDA approved |
|
|
Multiple cancers |
Phase I-III |
[52] |
||
|
Tremelimumab (Imjudo) |
Antineoplastic; liver cancer |
FDA approved |
||
|
PD-1 |
Nivolumab (Opdivo) |
Melanoma (2014), NSCLC (2015), and Renal (2018) cancers, Hodgkin lymphoma |
FDA approved |
|
|
Multiple cancers |
Phase I-III |
[52] |
||
|
Pembrolizumab (Keytruda) |
Melanoma (2014), Various (2015) |
FDA approved |
[56] |
|
|
Multiple cancers |
Phase I-III |
[52] |
||
|
MED10680 |
Multiple cancers |
Phase I |
[52] |
|
|
AMP-224 |
Multiple cancers |
Phase I |
[52] |
|
|
Pidilizumab |
Multiple cancers |
Phase I-II |
[52] |
|
|
Cemiplimab (Libtayo) |
Cutaneous squamous-cell carcinoma (2018) |
FDA approved |
||
|
PD-L1 |
Atezolizumab (Tecentriq) |
Bladder, NSCLC (2016), and TNBC (2019), hepatocellular carcinoma, HCC (2020) |
FDA approved |
[52] |
|
Avelumab (Bavencio) |
Urothelial Carcinoma (2017), Merkel Cell Carcinoma (2017), Renal carcinoma (2019) |
FDA approved |
[57] |
|
|
MED14736 |
Multiple cancers |
Phase III |
[52] |
|
|
Avelumab (Bavencio) |
Merkel cell carcinoma (2017), Rena (2019), Urothelial carcinoma (2020) |
FDA approved |
[57] |
|
|
BMS-936559 |
Multiple cancers |
Phase I |
[52] |
|
|
Durvalumab (IMFINZI) |
Bladder Cancer (2017), NSCLC (2018) |
FDA approved |
||
|
LAG-3 |
IMP321 |
Multiple cancers |
Phase I |
[52] |
|
BMS-986016 |
Multiple cancers |
Phase I |
[52] |
|
|
Relatlimab (Opdualag) |
Melanoma (2022) |
FDA approved |
[53] |
|
|
VEGF |
Bevacizumab |
Colorectal (2004), NSCLC (2006, 2018), Renal (2009), Cervical (2014), Glioblastoma (2009), and Ovarian (2018) Cancers |
FDA approved |
|
|
VEGF-A, Ang-2 |
Faricimab (Vabysmo) |
wAMD, DME |
FDA approved |
[53] |
|
VEGFR2 |
Ramucirumab (Cyramza) |
Gastric cancer (2014), NSCLC (2020), HCC (2019) |
FDA approved |
|
|
EGFR |
Cetuximab |
Colorectal cancer (CRC) (2004, 2012) and Head and neck squamous cell carcinoma (2006, 2011) |
FDA approved |
|
|
Necitumumab (Portrazza) |
NSCLC (2015) |
FDA approved |
||
|
Panitumumab (Vectibix) |
Colorectal Cancer (2006) |
FDA approved |
[59] |
|
|
PDGFRα |
Olaratumab (Lartruvo) |
Soft-tissue sarcoma (2016) |
FDA approved |
[59] |
|
HER2 |
Pertuzumab (Perjeta) |
HER2-positive Breast cancer (2012) |
FDA approved |
|
|
Trastuzumab (Herceptin) |
HER2-positive Breast cancer (1998) |
FDA approved |
||
|
Ado-trastuzumab emtansine (Kadcyla) |
HER2-Breast cancer (2013) |
FDA approved |
[59] |
|
|
Fam-trastuzumab deruxtecan (Enhertu) |
HER2-positive Breast cancer (2019) |
FDA approved |
[59] |
|
|
Trastuzumab tucatinib |
HER2-positive Breast cancer (2020) |
FDA approved |
[57] |
|
|
CCR4 |
Mogamulizumab (Poteligeo) |
Cutaneous T cell lymphoma (2018) |
FDA approved |
[59] |
|
CD20 |
Obinutuzumab (Gazyva) |
Chronic lymphocytic leukemia (2013), follicular lymphoma (2017) |
FDA approved |
|
|
Ofatumumab (Arzerra) |
Chronic lymphocytic leukemia (2014) |
FDA approved |
||
|
Rituximab (MabThera, Rituxan) |
B-Cell Lymphoma (1997) |
FDA approved |
[59] |
|
|
Ibritumomab tiuxetan (Zevalin) |
NHL (2004) |
FDA approved |
||
|
tositumomab Iodine-131 (Bexxar) |
NHL (2003) |
FDA approved |
[59] |
|
|
Ublituximab |
Chronic lymphocytic leukemia, CLL, non-Hodgkin’s lymphoma) and non-cancer (multiple sclerosis) |
Phase III |
||
|
CD33 |
Gemtuzumab ozogamicin (Mylotarg) |
Acute myeloid leukemia (2000) |
FDA approved |
|
|
CD30 |
Brentuximab vedotin (Adcetris) |
Hodgkin’s lymphoma and Anaplastic large-cell lymphoma (2011) |
FDA approved |
|
|
CD79B |
Polatuzumab vedotin (Polivy) |
Diffuse large B-cell lymphoma (2019) |
FDA approved |
|
|
CD22 |
Inotuzumab ozogamicin (BESPONSA) |
Acute lymphoblastic leukemia (2017) |
FDA approved |
|
|
Moxetumomab pasudotox (Lumoxiti) |
Hairy-cell leukemia (2018) |
FDA approved |
||
|
CD19 |
Inebilizumab (Uplizna) |
Neuromyelitis optica and neuromyelitis optica spectrum disorders (2022) |
FDA approved |
[53] |
|
CD19, CD3 |
Blinatumomab (Blincyto) |
Acute lymphoblastic leukemia (2014) |
FDA approved |
|
|
TROP2 |
Sacituzumab govitecan (Trodelvy) |
TNBC (2020) |
FDA approved |
|
|
CD3 |
Muromonab-CD3 (Orthoclone Okt3) |
Reversal of kidney transplant rejection (1986) |
FDA approved |
[60] |
|
CD3, BCMA |
Teclistamab (TECVAYLI) |
Multiple myeloma (2022) |
FDA approved |
[53] |
|
gp100, CD3 |
Tebentafusp (KIMMTRAK) |
Metastatic uveal melanoma (2022) |
FDA approved |
[53] |
|
CD30, CD3 |
Mosunetuzumab (Lunsumio) |
Follicular lymphoma (2022) |
FDA Review |
[53] |
|
CD38 |
Daratumumab (Darzalex) |
Multiple Myeloma (2015) |
FDA approved |
|
|
Isatuximab (Sarclisa) |
Multiple Myeloma (2020) |
FDA approved |
||
|
GD2 |
Dinutuximab (Qarziba; Unituxin) |
Neuroblastoma (2015) |
FDA approved |
|
|
Nectin-4 |
Enfortumab Vedotin (Padcev) |
Bladder cancer (2019), Urothelial cancer (2022) |
FDA approved |
|
|
Small Drugs Based |
||||
|---|---|---|---|---|
|
Target |
Drug INN (Brand Name) |
Cancer Type |
Current Status |
Ref. |
|
EGFR |
Gefitinib |
NSCLC (2015) |
FDA approved |
|
|
Erlotinib HCl (Tarceva) |
NSCLC (2004) |
FDA approved |
[63] |
|
|
Osimertinib mesylate |
NSCLC (2020) |
FDA approved |
||
|
Dacomitinib (Vizimpro) |
EGFR-mutated NSCLC (2018) |
FDA approved |
||
|
Mobocertinib succinate (Exkivity) |
EGFR exon 20-mutated NSCLC (2021) |
FDA approved |
[63] |
|
|
HER2 |
Tucatinib (Tukysa) |
HER2-positive breast cancer (2020) |
FDA approved |
|
|
EGFR, HER2, and HER4 |
Neratinib maleate (Nerlynx) |
HER2-overexpressed breast cancer (2017) |
FDA approved |
[63] |
|
Afatinib dimaleate (Gilotrif) |
Metastatic NSCLC with EGFR exon 19 deletion or exon 21 (L858R) mutation (2013) |
FDA approved |
[63] |
|
|
PARP |
Olaparib (Lynparza) |
Advanced BRCA-mutated ovarian cancer (2020) |
FDA approved |
|
|
Rucaparib camsylate (Rubraca) |
BRCA-positive ovarian cancer (2016) |
FDA approved |
||
|
Niraparib tosylate (Zejula) |
Epithelial ovarian, fallopian tube, or primary peritoneal cancer (2017) |
FDA approved |
[63] |
|
|
PDGFRα |
Avapritinib (Ayvakit) |
metastatic gastrointestinal stromal tumor (GIST) with platelet-derived growth factor receptor alpha (PDGFRA) exon 18 mutations (2020) |
FDA approved |
|
|
Multitarget TKI (VEGFRs, PDGFRα/β, CSF1R, KIT, and FLT3) |
Sunitinib malate (Sutent) |
Imatinib-resistant GIST and advanced RCC (2013) |
FDA approved |
|
|
Multitarget TKI (RET, VEGFRs, KIT, PDGFRα/β, FGFR1/2, RAF1, BRAF, and BRAFV600E) |
Regorafenib (Stivarga) |
Metastatic colorectal cancer (2012) |
FDA approved |
|
|
Multitarget TKI (VEGFR2/3, PDGFRβ, FLT3, KIT, RAF1, and BRAF) |
Sorafenib toylate (Nexavar) |
Advanced RCC (2005) |
FDA approved |
[63] |
|
Multitarget TKI (VEGFRs, PDGFRα/β, FGFR1/2, KIT) |
Pazopanib HCl (Votrient) |
Metastatic RCC (2009) |
FDA approved |
[63] |
|
Multitarget TKI (VEGFRs, FGFRs, PDGFRα, RET, and KIT) |
Lenvatinib mesylate (Lenvima) |
Thyroid cancer (2015) |
FDA approved |
[63] |
|
Multitarget TKI (VEGFR2/3, EGFR, and RET) |
Vandetanib (Caprelsa) |
Unresectable or metastatic medullary thyroid cancer (2011) |
FDA approved |
|
|
Multitarget TKI (VEGFRs, MET, RET, FLT3, KIT, TIE2, and AXL) |
Cabozantinib S-malate (Cometriq) |
Progressive, metastatic medullary thyroid cancer (2012) |
[63] |
|
|
VEGFRs |
Axitinib (Inlyta) |
Advanced RCC (2012) |
FDA approved |
|
|
Tivozanib HCl (Fotivda) |
Advanced RCC (2021) |
FDA approved |
[63] |
|
|
mTOR |
Everolimus (Afinitor) |
Advanced RCC (2009), HER2-negative breast cancer after failure of treatment with letrozole or anastrozole (2012), nonfunctional neuroendocrine tumors of gastrointestinal or lung origin with unresectable, locally advanced, or metastatic disease (2016) |
FDA approved |
|
|
Temsirolimus (Torisel) |
Advanced RCC (2007) |
FDA approved |
[63] |
|
This entry is adapted from the peer-reviewed paper 10.3390/vaccines11010059
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33.
- Zeng, D.; Li, M.; Zhou, R.; Zhang, J.; Sun, H.; Shi, M.; Bin, J.; Liao, Y.; Rao, J.; Liao, W. Tumor Microenvironment Characterization in Gastric Cancer Identifies Prognostic and Immunotherapeutically Relevant Gene Signatures. Cancer Immunol. Res. 2019, 7, 737–750.
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437.
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416.
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598.
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734.
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682.
- Christiansen, A.; Detmar, M. Lymphangiogenesis and cancer. Genes Cancer 2011, 2, 1146–1158.
- Fukumura, D.; Xavier, R.; Sugiura, T.; Chen, Y.; Park, E.C.; Lu, N.; Selig, M.; Nielsen, G.; Taksir, T.; Jain, R.K.; et al. Tumor induction of VEGF promoter activity in stromal cells. Cell 1998, 94, 715–725.
- Joukov, V.; Pajusola, K.; Kaipainen, A.; Chilov, D.; Lahtinen, I.; Kukk, E.; Saksela, O.; Kalkkinen, N.; Alitalo, K. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. Embo J. 1996, 15, 290–298.
- Orlandini, M.; Marconcini, L.; Ferruzzi, R.; Oliviero, S. Identification of a c-fos-induced gene that is related to the platelet-derived growth factor/vascular endothelial growth factor family. Proc. Natl. Acad. Sci. USA 1996, 93, 11675–11680.
- Yamada, Y.; Nezu, J.; Shimane, M.; Hirata, Y. Molecular cloning of a novel vascular endothelial growth factor, VEGF-D. Genomics 1997, 42, 483–488.
- Achen, M.G.; Jeltsch, M.; Kukk, E.; Mäkinen, T.; Vitali, A.; Wilks, A.F.; Alitalo, K.; Stacker, S.A. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc. Natl. Acad. Sci. USA 1998, 95, 548–553.
- Favier, B.; Alam, A.; Barron, P.; Bonnin, J.; Laboudie, P.; Fons, P.; Mandron, M.; Herault, J.P.; Neufeld, G.; Savi, P.; et al. Neuropilin-2 interacts with VEGFR-2 and VEGFR-3 and promotes human endothelial cell survival and migration. Blood 2006, 108, 1243–1250.
- Soker, S.; Miao, H.Q.; Nomi, M.; Takashima, S.; Klagsbrun, M. VEGF165 mediates formation of complexes containing VEGFR-2 and neuropilin-1 that enhance VEGF165-receptor binding. J. Cell Biochem. 2002, 85, 357–368.
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56.
- Hirakawa, S.; Detmar, M.; Kerjaschki, D.; Nagamatsu, S.; Matsuo, K.; Tanemura, A.; Kamata, N.; Higashikawa, K.; Okazaki, H.; Kameda, K.; et al. Nodal lymphangiogenesis and metastasis: Role of tumor-induced lymphatic vessel activation in extramammary Paget’s disease. Am. J. Pathol. 2009, 175, 2235–2248.
- Zhang, J.P.; Lu, W.G.; Ye, F.; Chen, H.Z.; Zhou, C.Y.; Xie, X. Study on CXCR4/SDF-1alpha axis in lymph node metastasis of cervical squamous cell carcinoma. Int. J. Gynecol. Cancer 2007, 17, 478–483.
- Kaifi, J.T.; Yekebas, E.F.; Schurr, P.; Obonyo, D.; Wachowiak, R.; Busch, P.; Heinecke, A.; Pantel, K.; Izbicki, J.R. Tumor-cell homing to lymph nodes and bone marrow and CXCR4 expression in esophageal cancer. J. Natl. Cancer Inst. 2005, 97, 1840–1847.
- Yoshitake, N.; Fukui, H.; Yamagishi, H.; Sekikawa, A.; Fujii, S.; Tomita, S.; Ichikawa, K.; Imura, J.; Hiraishi, H.; Fujimori, T. Expression of SDF-1 alpha and nuclear CXCR4 predicts lymph node metastasis in colorectal cancer. Br. J. Cancer 2008, 98, 1682–1689.
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51.
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330.
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80.
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567.
- Bai, R.; Lv, Z.; Xu, D.; Cui, J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark Res. 2020, 8, 34.
- Woo, S.R.; Corrales, L.; Gajewski, T.F. Innate immune recognition of cancer. Annu. Rev. Immunol. 2015, 33, 445–474.
- Pagès, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N. Engl. J. Med. 2005, 353, 2654–2666.
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964.
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949.
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022.
- Liu, R.B.; Engels, B.; Arina, A.; Schreiber, K.; Hyjek, E.; Schietinger, A.; Binder, D.C.; Butz, E.; Krausz, T.; Rowley, D.A.; et al. Densely granulated murine NK cells eradicate large solid tumors. Cancer Res. 2012, 72, 1964–1974.
- Delahaye, N.F.; Rusakiewicz, S.; Martins, I.; Ménard, C.; Roux, S.; Lyonnet, L.; Paul, P.; Sarabi, M.; Chaput, N.; Semeraro, M.; et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat. Med. 2011, 17, 700–707.
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl. Med. 2017, 9, eaal3604.
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tillé, L.; Homicsko, K.; Damsky, W.; Maby-El Hajjami, H.; et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, eaan3311.
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e16.
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757.
- Crist, M.; Yaniv, B.; Palackdharry, S.; Lehn, M.A.; Medvedovic, M.; Stone, T.; Gulati, S.; Karivedu, V.; Borchers, M.; Fuhrman, B.; et al. Metformin increases natural killer cell functions in head and neck squamous cell carcinoma through CXCL1 inhibition. J. Immunother. Cancer 2022, 10, e005632.
- Wang, S.; Lin, Y.; Xiong, X.; Wang, L.; Guo, Y.; Chen, Y.; Chen, S.; Wang, G.; Lin, P.; Chen, H.; et al. Low-Dose Metformin Reprograms the Tumor Immune Microenvironment in Human Esophageal Cancer: Results of a Phase II Clinical Trial. Clin. Cancer Res. 2020, 26, 4921–4932.
- Wei, Z.; Zhang, X.; Yong, T.; Bie, N.; Zhan, G.; Li, X.; Liang, Q.; Li, J.; Yu, J.; Huang, G.; et al. Boosting anti-PD-1 therapy with metformin-loaded macrophage-derived microparticles. Nat. Commun. 2021, 12, 440.
- Liu, Y.; Zhou, Z.; Hou, J.; Xiong, W.; Kim, H.; Chen, J.; Zheng, C.; Jiang, X.; Yoon, J.; Shen, J. Tumor Selective Metabolic Reprogramming as a Prospective PD-L1 Depression Strategy to Reactivate Immunotherapy. Adv. Mater. 2022, 34, e2206121.
- Chen, J.; Zhou, Z.; Zheng, C.; Liu, Y.; Hao, R.; Ji, X.; Xi, Q.; Shen, J.; Li, Z. Chitosan oligosaccharide regulates AMPK and STAT1 pathways synergistically to mediate PD-L1 expression for cancer chemoimmunotherapy. Carbohydr. Polym. 2022, 277, 118869.
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668.
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182.
- Walker, L.S. Treg and CTLA-4: Two intertwining pathways to immune tolerance. J. Autoimmun. 2013, 45, 49–57.
- Chikuma, S.; Bluestone, J.A. Expression of CTLA-4 and FOXP3 in cis protects from lethal lymphoproliferative disease. Eur. J. Immunol. 2007, 37, 1285–1289.
- Van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647.
- Traversari, C.; van der Bruggen, P.; Luescher, I.F.; Lurquin, C.; Chomez, P.; Van Pel, A.; De Plaen, E.; Amar-Costesec, A.; Boon, T. A nonapeptide encoded by human gene MAGE-1 is recognized on HLA-A1 by cytolytic T lymphocytes directed against tumor antigen MZ2-E. J. Exp. Med. 1992, 176, 1453–1457.
- Hanna, M.G., Jr.; Peters, L.C. Immunotherapy of established micrometastases with Bacillus Calmette-Guérin tumor cell vaccine. Cancer Res. 1978, 38, 204–209.
- Guo, C.; Manjili, M.H.; Subjeck, J.R.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. Therapeutic cancer vaccines: Past, present, and future. Adv. Cancer Res. 2013, 119, 421–475.
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337.
- Barbee, M.S.; Ogunniyi, A.; Horvat, T.Z.; Dang, T.O. Current status and future directions of the immune checkpoint inhibitors ipilimumab, pembrolizumab, and nivolumab in oncology. Ann. Pharm. 2015, 49, 907–937.
- Zhang, H.; Chen, J. Current status and future directions of cancer immunotherapy. J. Cancer 2018, 9, 1773–1781.
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat. Rev. Drug. Discov. 2021, 20, 491–495.
- Marrone, K.A.; Brahmer, J.R. Using Immune Checkpoint Inhibitors in Lung Cancer. Oncology 2016, 30, 713–721.
- Hatae, R.; Chamoto, K. Immune checkpoint inhibitors targeting programmed cell death-1 (PD-1) in cancer therapy. Rinsho Ketsueki 2016, 57, 2224–2231.
- Swart, M.; Verbrugge, I.; Beltman, J.B. Combination Approaches with Immune-Checkpoint Blockade in Cancer Therapy. Front. Oncol. 2016, 6, 233.
- Olivier, T.; Haslam, A.; Prasad, V. Anticancer Drugs Approved by the US Food and Drug Administration From 2009 to 2020 According to Their Mechanism of Action. JAMA Netw. Open 2021, 4, e2138793.
- Kates, M.; Sopko, N.A.; Matsui, H.; Drake, C.G.; Hahn, N.M.; Bivalacqua, T.J. Immune checkpoint inhibitors: A new frontier in bladder cancer. World J. Urol. 2016, 34, 49–55.
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34.
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1.
- Chen, R.; Chen, B. Brentuximab vedotin for relapsed or refractory Hodgkin’s lymphoma. Drug. Des. Devel. 2015, 9, 1729–1733.
- Chen, R.; Hou, J.; Newman, E.; Kim, Y.; Donohue, C.; Liu, X.; Thomas, S.H.; Forman, S.J.; Kane, S.E. CD30 Downregulation, MMAE Resistance, and MDR1 Upregulation Are All Associated with Resistance to Brentuximab Vedotin. Mol. Cancer 2015, 14, 1376–1384.
- Wu, Q.; Qian, W.; Sun, X.; Jiang, S. Small-molecule inhibitors, immune checkpoint inhibitors, and more: FDA-approved novel therapeutic drugs for solid tumors from 1991 to 2021. J. Hematol. Oncol. 2022, 15, 143.