Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Allergy
Systemic lupus erythematosus (SLE) is a chronic idiopathic autoimmune disease affecting various organs including the central nervous system, presenting as seizures and psychosis. Patients with SLE also present numerous neuropsychiatric manifestations. These neuropsychiatric manifestations are referred to as neuropsychiatric systemic lupus erythematosus (NPSLE). NPSLE affects both the central nervous system (CNS) and the peripheral nervous system (PNS) and can present as various symptoms, such as cognitive dysfunction, organic brain syndromes, delirium, seizures, headache, and psychosis [1].
Some people with NPSLE have problems with their brain and nerves. They may have strokes, confusion, mood disorders, and trouble with thinking and memory. This is called NPSLE, and it affects from 3 to 4 out of 10 people with SLE. Sometimes, NPSLE is the first sign of SLE, and it can happen even when SLE is not very active. Scientists are trying to figure out what causes NPSLE. They think it may have to do with blood clots, antibodies that attack the brain, and proteins that cause inflammation. They also want to know how some chemicals in the body, like TNF, IL-1, IL-6, and IFN-γ, affect the brain and nerves. Some new studies suggest that another chemical, called TWEAK, may play a role in NPSLE in humans and mice [2].
In SLE there are eight types of pleuropulmonary involvement: lupus pleuritis, acute lupus pneumonitis, pleural effusion, shrinking lung syndrome, diffuse alveolar hemorrhage, pulmonary arterial hypertension, interstitial lung disease, and pulmonary embolism [3].
Likewise, the skin is mostly affected by “butterfly rash”, photosensitivity, and vasculitis [4]. SLE affects the heart, including pericarditis and endocarditis. Glomerulonephritis is a renal involvement of SLE [5], as well as diseases of the joints such as arthritis [6,7]. Lymphadenopathy, autoimmune haemolytic anaemia, autoimmune leukopenia, and autoimmune thrombocytopenia [8] are also manifestations of SLE.
When cells fail to die properly, they can trigger autoimmune disease. A faulty ‘death receptor’ called FAS prevents the removal of self-reactive lymphocytes in the body, causing them to accumulate and cause inflammation and organ damage in mice (Fas(lpr/lpr) (mice with a genetic mutation that causes excessive cell growth) and humans. The REL/NF-κB family of proteins controls many aspects of immunity and is also involved in different aspects of autoimmunity [9]. Recurrent infections such as pneumonias [10,11], oral ulcers [12], and discoid lupus are present in numerous individuals with SLE. Gastrointestinal manifestations include lupus mesenteric vasculitis, intestinal pseudo-obstruction, and protein-losing enteropathy [13].
Notably, the presence of neuropsychiatric symptoms (NPS) in SLE patients does not explicitly indicate the cause of SLE. This is because NPS can be comorbid, coincidental, or a complication of SLE treatment, most notably psychotropic drugs such as corticosteroids. NPSLE is further classified as either primary or secondary [1]. Primary NPSLE syndromes result from direct CNS autoimmune inflammatory processes, whereas secondary NPSLE syndromes are caused by indirect complications of SLE such as treatment side effects, CNS infection from chronic immunosuppression, or SLE-related organ damage [14].
- Molecules, Neurosychiatric systemic lupus erythematosus, cytokines, autoantibodies, brain
1. Introduction
Systemic lupus erythematosus (SLE) is a chronic idiopathic autoimmune disease affecting various organs including the central nervous system, presenting as seizures and psychosis. Patients with SLE also present numerous neuropsychiatric manifestations. These neuropsychiatric manifestations are referred to as neuropsychiatric systemic lupus erythematosus (NPSLE). NPSLE affects both the central nervous system (CNS) and the peripheral nervous system (PNS) and can present as various symptoms, such as cognitive dysfunction, organic brain syndromes, delirium, seizures, headache, and psychosis [1].
Some people with NPSLE have problems with their brain and nerves. They may have strokes, confusion, mood disorders, and trouble with thinking and memory. This is called NPSLE, and it affects from 3 to 4 out of 10 people with SLE. Sometimes, NPSLE is the first sign of SLE, and it can happen even when SLE is not very active. Scientists are trying to figure out what causes NPSLE. They think it may have to do with blood clots, antibodies that attack the brain, and proteins that cause inflammation. They also want to know how some chemicals in the body, like TNF, IL-1, IL-6, and IFN-γ, affect the brain and nerves. Some new studies suggest that another chemical, called TWEAK, may play a role in NPSLE in humans and mice [2].
In SLE there are eight types of pleuropulmonary involvement: lupus pleuritis, acute lupus pneumonitis, pleural effusion, shrinking lung syndrome, diffuse alveolar hemorrhage, pulmonary arterial hypertension, interstitial lung disease, and pulmonary embolism [3].
Likewise, the skin is mostly affected by “butterfly rash”, photosensitivity, and vasculitis [4]. SLE affects the heart, including pericarditis and endocarditis. Glomerulonephritis is a renal involvement of SLE [5], as well as diseases of the joints such as arthritis [6,7]. Lymphadenopathy, autoimmune haemolytic anaemia, autoimmune leukopenia, and autoimmune thrombocytopenia [8] are also manifestations of SLE.
When cells fail to die properly, they can trigger autoimmune disease. A faulty ‘death receptor’ called FAS prevents the removal of self-reactive lymphocytes in the body, causing them to accumulate and cause inflammation and organ damage in mice (Fas(lpr/lpr) (mice with a genetic mutation that causes excessive cell growth) and humans. The REL/NF-κB family of proteins controls many aspects of immunity and is also involved in different aspects of autoimmunity [9]. Recurrent infections such as pneumonias [10,11], oral ulcers [12], and discoid lupus are present in numerous individuals with SLE. Gastrointestinal manifestations include lupus mesenteric vasculitis, intestinal pseudo-obstruction, and protein-losing enteropathy [13].
Notably, the presence of neuropsychiatric symptoms (NPS) in SLE patients does not explicitly indicate the cause of SLE. This is because NPS can be comorbid, coincidental, or a complication of SLE treatment, most notably psychotropic drugs such as corticosteroids. NPSLE is further classified as either primary or secondary [1]. Primary NPSLE syndromes result from direct CNS autoimmune inflammatory processes, whereas secondary NPSLE syndromes are caused by indirect complications of SLE such as treatment side effects, CNS infection from chronic immunosuppression, or SLE-related organ damage [14].
Psychosis is an uncommon neuropsychiatric manifestation of NPSLE but can be secondary to long-term, high-dose glucocorticoids. Glucocorticoids are among the mainstay drugs for the treatment of NPSLE, which makes management even more difficult as they are known to have psychiatric side effects and can cause a spectrum of psychiatric symptoms, including mania, psychosis, anxiety, and depression. On initial clinical examination, it may be difficult to differentiate the cause of psychosis as a result of steroids or NPSLE because no single laboratory test is currently available to definitively confirm the diagnosis of NPSLE [15].
There are 19 neuropsychiatric syndromes observed in SLE, as listed by the American College of Rheumatology (ACR) Nomenclature for NPSLE [16]. The ACR’s nomenclature for NPSLE was last revisited in 2021 [12], as shown in Table 1. NPSLE can precede the onset of lupus or occur at any time during its course [17].
Table 1. Clinical syndromes in NPSLE. Taken from [18].
| Central Nervous System | Neurological syndromes (focal): Seizure disorder Aseptic meningitis Cerebrovascular disease Demyelinating syndromes Headache Myelopathy Movement disorders Neuropsychiatric syndrome (diffuse): Anxiety disorders Psychosis Mood disorders Acute confusional state Cognitive dysfunction |
| Peripheral Central Nervous System | Neurological syndromes (focal): Autonomic disorders Myasthenia gravis Polyneuropathy Guillian Barre Syndrome Plexopathy Mononeuropathy |
Table 1 shows that, although the frequency of NPSLE syndromes varies tremendously, they could be used as qualitative diagnostic criteria if present to confirm the diagnosis of NPSLE. In addition, this should be accompanied by some laboratory findings for SLE, including the presence of antinuclear antibodies (ANA) such as anti-ds DNA antibodies, which are the hallmark of SLE [17,19].
2. Most Systemic Lesions Are Due to Loss of Tolerance to Self-Antigens
The exact etiology of SLE remains to be fully delineated; however, most systemic lesions are due to the loss of tolerance to self-antigens, including histone, ribonucleoprotein, double-stranded DNA, antigen Ro (SSA), and antigen La (SSB); direct or indirect damage from autoantibody formation; and the generation of immune complexes (type III hypersensitivity) [20]. This was confirmed by the fact that anti-DNA complexes can be found in many organs such as the kidneys, lungs, and blood vessels. Serum complement levels, which are also measured during the initial workup and disease monitoring, can markedly decrease secondary to consumption and granular deposition. Therefore, low serum complement levels and immunofluorescence which illustrates granular complement deposits in the glomeruli further support the immunological etiology of the disease.
Alterations in B and T cell activation, along with an impaired clearance of apoptotic debris, have also been implicated in SLE histopathology. NPSLE patients showed significantly more microinfarction, macroinfarction, vasculitis, and microthrombi upon histological analysis than SLE patients without neuropsychiatric manifestations [21]. The histopathological analysis of NPSLE patients varies from nonspecific findings of focal vasculopathy to more specific lesions, including C4d- and C5b-9-associated microthrombi and diffuse vasculopathy [22], which often correlate with the clinical syndromes that define NPSLE [23].
3. SLE Immunopathogenesis
3.1. Apoptosis Cascade and Role of IFN-α in SLE
A low clearance of apoptotic debris in SLE will lead to an increased formation of autoantigen-antibody complexes by autoreactive B cells. Initially, granulocytes and other innate cells, via pattern-recognition receptors, will recognize apoptotic bodies. The binding of ligands with toll-like receptors (TLRs) activates B-cells, which produce autoantibodies leading to immune complex formation and the activation of the complement system. Plasmacytoid dendritic cells (pDC) stimulate the synthesis of endogenous type I interferon (IFN-α) via a TLR7- and TLR9-dependent pathway. This causes the synthesis of cytokines including (IL)-1β and IL-23 and stimulates Th17 differentiation [24]. Figure 1 shows the type III hypersensitivity mechanism (immunocomplex-mediated) which is involved in the immunopathogenesis of SLE.
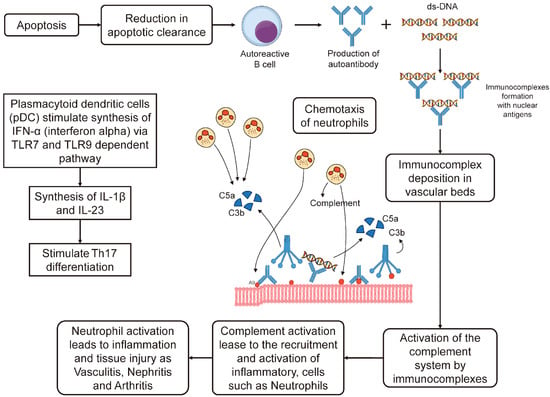
Figure 1. In Systemic lupus erythematosus (SLE), a type III hypersensitivity reaction occurs. This involves the formation of immune complexes that trigger the activation of the complement system. Two components of this system, C3a and C5a, serve as chemotactic factors. They draw neutrophils to the location where the immune complexes are deposited. The activation of these neutrophils results in inflammation and damage to the site, leading to conditions such as vasculitis, nephritis, and arthritis. There may also be other mechanisms at play. Adapted from [24].
3.2. Increased Association of HLA System and SLE in a Population
It was found, in the Saudi population, that some HLA alleles increased the risk of SLE: HLA-A29 (OR = 2.70; 95% CI = 1.03–7.08; p = 0.0035), HLA-B51 (OR = 1.81; 95% CI = 1.17–2.79; p = 0.0066), HLA-DRB115 (OR = 1.45; 95% CI = 0.98–2.29; p = 0.063), and HLA-DQB106 (OR = 1.67; 95% CI = 1.19–2.36; p = 0.0032). On the other hand, HLA-DRB116 reduced the likelihood of the disease (OR = 0.18; 95% CI = 0.02–1.3; p = 0.055). HLA-DRB115 haplotypes had a significant link with SLE (OR = 2.01, 95% CI = 1.20–3.68, p = 0.008), mainly because of the HLADRB115–DQB106 combination. The results indicate a relationship between class I and class II MHC (HLA-A29, HLA-B51, HLA-DRB115, and HLA-DQB106) and SLE vulnerability in the Saudi population [25].
3.3. Immunopathogenesis of NPSLE
3.3.1. Genetic Factors
The immunopathogenesis of NPSLE is shown in Figure 2. It is shown to include genetic factors such as the TREX1 gene and HLA-DRB1*04 [12]. The TREX1 gene encodes for TREX1, which is a protein that is expressed extensively and functions as a component of the SET complex in the process of granzyme A-mediated apoptosis, where it degrades single-stranded DNA. The TREX1 gene codes for a 3′-exonuclease 1 protein, which eliminates nucleotides from the 3′ ends of DNA strands, thereby removing unnecessary fragments that might have been produced during DNA replication. The TREX1 gene has been identified to have a role in the regulation of the immune system and in viral infections. Studies have discovered that alterations in this gene are associated with numerous diseases, such as Aicardi-Goutieres syndrome (AGS), systemic lupus erythematosus (SLE), familial chilblain lupus (FCL), retinal vasculopathy, and cerebral leukodystrophy. A common feature in these autoimmune diseases is the frequent detection of antibodies to double-stranded DNA (dsDNA) [26,27,28]. The homodimer TREX1R114H/R114H exhibits impaired dsDNA and ssDNA degradation functions and does not noticeably hinder the TREX1WT enzyme. On the other hand, the heterodimer TREX1WT/R114H retains functional dsDNA degradation activity, which corroborates the recessive genetic nature of TREX1 R114H in AGS and the proposed mechanism of the TREX1 exonuclease [28].
As previously mentioned, among the genetic factors for NPSLE, HLA-DRB1*04 could play a critical role. In Malay SLE patients, HLA-DRB1*04 was found to have a significant correlation with lupus nephritis, characterized by high levels of anti-ds DNA Ab, and arthritis. Further analysis of the association of HLA-DRB1*04 with clinical and biological factors showed that it had a significant correlation with the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) scores, C-reactive protein (CRP) in the blood, anti-nuclear antibody (ANA), and total protein in the urine. It was also observed that SLE carriers possessing the HLA-DRB1*04 allele had a significant correlation with elevated levels of cytokines such as IL-17F and GM-CSF [29].
In summary, some the genetic factors involved in SLE are:
-
Transcriptomic data analysis has revealed several pathways and immune responses that are associated with SLE, such as interferons, T cell differentiation, complement pathways, and coagulation;
-
Eight genes (SOCE, CXCL8, MMP9, IL1B, JUN, TNF, NFKBIA, and FOS) are up-regulated in SLE and have interactions with different pathways. These genes are also linked to SNPs that are identified by GWAS;
-
Several other genes with known SLE-related variations are detected by integrating GWAS and pathway analysis, such as TYK2, SH2B, C5, IL2RA, IRF5, FCGR2A, TNFAIP3, STAT4, LYN, IL7R, and HLA-DRB;
-
One of the relevant pathways that is identified by pathway-based analysis is the TSLP signaling pathway, which is connected to rs7574865, LYN, STAT4, and IL7R;
3.3.2. Comorbidities
Lupus nephritis (LN) and NPSLE are both severe manifestations of SLE, a chronic autoimmune disease [33]. LN is a type of glomerulonephritis mediated by immune complex deposition at glomerular sites, representing one of the severe major organ involvements seen in SLE. On the other hand, NPSLE refers to a series of neurological and psychiatric symptoms directly related to SLE that involve the central and peripheral nervous system [33].
The comorbidity of LN and NPSLE in SLE patients can be explained by the systemic nature of SLE, which can affect any organ, including the kidneys and the nervous system [34]. The immune response in SLE patients can lead to inflammation and damage in various parts of the body, including the kidneys (leading to LN) and the nervous system (leading to NPSLE) [29].
Moreover, neuropsychiatric symptoms, whether causally associated or comorbid, negatively impact the quality of life of patients with SLE. In addition, these symptoms appear to identify patients with a higher mortality than those without neuropsychiatric symptoms. It is important to note that, while the attribution of neurologic symptoms to SLE may influence decisions about disease-modifying treatments, the timely recognition of neuropsychiatric comorbidity in SLE patients is also important to provide appropriate symptomatic management [34]. Therefore, understanding the comorbidity of LN and NPSLE in SLE patients is crucial for their management and treatment.
The clinical manifestations depend on environmental, immunological, hormonal, and genetic factors. The blood–brain barrier (BBB) can breach through multiple mechanisms. These include autoimmune processes, such as immune complex deposition and pathologic cytokine-mediated destruction, and environmental factors, such as smoking and hypertension [35]. There are no specific abnormalities noted on the brain images of patients with NPSLE, and some may even have normal findings, nonspecific white matter changes, or atrophy [14].
3.3.3. Summary of NPSLE Immunopathogenesis
In summary, NPSLE is a complex condition with a multifaceted pathogenesis involving genetic factors, cytokines, immune cells, and environmental factors [36], as shown in Figure 2.
Genetic Factors: Certain genes have been associated with the development of NPSLE. For instance, specific alleles of the human leukocyte antigen (HLA) genes have been linked to an increased risk of developing NPSLE [36,37,38]. Researchers already refer to the TREX1 gene as a genetic factor in the immunopathogenesis of this autoimmune disorder [12].
Cytokines: Cytokines, which are signaling molecules in the immune system, play a crucial role in the pathogenesis of NPSLE. They mediate inflammation and immune responses, contributing to the neurological and psychiatric symptoms observed in NPSLE. For example, increased levels of certain cytokines (like IFN-y, IL-17F, IL-21, IL-18, GM-CSF, and VEGF) have been observed in SLE patients [36].
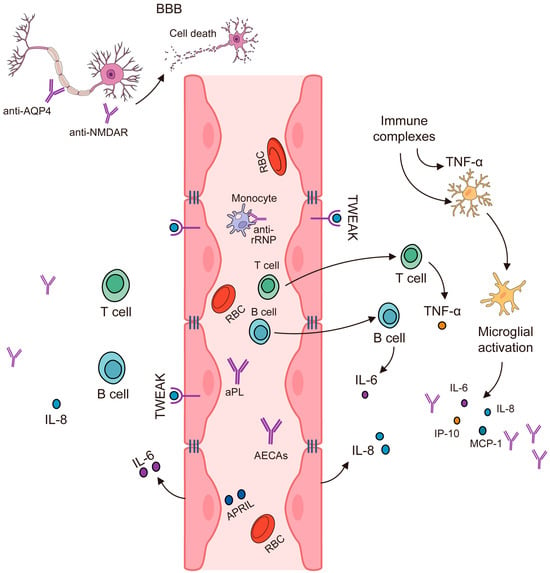
Figure 2. Immunopathogenesis of NPSLE. Modified from [39].
Immune Cells: The role of immune cells in NPSLE is significant. Autoantibodies produced by B cells can cross the blood–brain barrier (BBB) and bind to brain tissue, causing inflammation and damage [40]. T cells also contribute to the pathogenesis of NPSLE by producing pro-inflammatory cytokines [39], as shown in Figure 2.
Environmental Factors: Environmental factors such as infections, stress, smoking, and exposure to ultraviolet light can trigger the onset of NPSLE in genetically predisposed individuals [36].
Pathogenesis: The pathogenesis of NPSLE is thought to involve a combination of the above factors. The presence of certain autoantibodies (like anti-ribosomal P, anti-NR2, and anti-16/6 Id antibodies) and brain cytoplasmic ribonucleic acid (BC RNA) antibodies, which disrupt normal brain function, have been found in NPSLE patients [40,41]. These antibodies, along with the cytokines, can cause neurocognitive symptoms [18,36,41,42]. Additionally, the dysfunction of the BBB and vascular lesions may contribute to the development of NPSLE [36]. It is important to note that the exact mechanisms are still being researched, and the pathogenesis likely involves a complex interplay of these factors [36,39]. Other factors to take into consideration regarding the immunopathogenesis of NPSLE are the presence and interaction of chemokines with the innate and acquired immune system, the existence of lymphocytic infiltrate, the activation of endothelial cells in the brain, the activation of microglial cells, and neuronal cell death by apoptosis [42].
3.4. IL-2, IL-10, and IFN-γ Produced by T-Helper Cells Are Elevated in NPSLE
IL-2, IL-10, and IFN-γ produced by T-helper cells are elevated in NPSLE, and this is directly related to autoimmune and pro-inflammatory states [18,43]. The neuropsychiatric manifestations of SLE are likely due to antibodies that react with neurons either directly or indirectly via the activation of other neural cells and cross the BBB due to its disruption. Other immunological factors, such as cytokine-mediated CNS toxicity, may also play a role. Some of the cytokines found to be elevated in patients with NPSLE include IL-2, IL-10, and IFN-γ produced by T-helper cells [44]. The pathogenesis of compromised BBB integrity is not yet fully understood. However, once they enter the CNS, antibodies and cytokines can cause clinical effects. Therefore, clinically useful biomarkers must be identified [37].
3.5. Noninflammatory or Thrombotic/Ischemic Vascular Injury
Noninflammatory complications are associated with vascular thrombosis and hemorrhage. The thrombosis of large and small intracranial vessels can occur due to immune complex damage, antibody-mediated damage, complete deposition, accelerated atherosclerosis, or leucoagglutination [1,18,45]. Cerebral vasculitis has also been associated with NPSLE. CNS vasculopathy via antibodies, such as anti-phospholipid antibodies, may damage the BBB and allow CNS immune complex deposition, resulting in NPSLE. However, CNS inflammatory vasculopathy is rare, and non-inflammatory vasculopathy is more commonly observed [46].
While this classification is useful for descriptive purposes, both inflammatory and non-inflammatory complications can occur [47]. These manifestations of NPSLE are classified based on the 19 syndromes described by the ACR; however, they are not yet fully understood. The predominant inflammatory syndromes can result from the generation of pathological autoantibodies associated with cytokine-mediated damage [44].
4. Autoantibodies Can Lead to Neuronal Damage in NPSLE
An overriding feature of SLE is the involvement of the immune system and the production of autoantibodies. The immune mediators of NPSLE are quite extensive and include autoantibodies, cytokines, and chemokines. Autoantibodies can lead to neuronal damage and promote the pathogenesis of NPSLE. More than 116 antibodies have been reported for SLE, and at least 11 brain-specific and 9 systemic antibodies have been associated with NPSLE [36,48]. However, none of these autoantibodies have definitive implications in the pathogenesis of NPSLE, and their association remains controversial [49].
Brain-specific antibodies associated with NPSLE include anti-neuronal Abs, brain-reactive Abs (BRAA), Anti-N-methyl-D-aspartate receptor Abs (NMDA), anti-microtubule-associated protein 2 Abs (MAP-2), anti-neurofilament Abs (ANFA), anti-ganglioside Abs (AGA), anti-central nervous system tissue (CNS) Abs, anti-brain-synaptosomal Abs, anti-triosephosphate isomerase (TPI) Abs, anti-glial fibrillary acidic protein (GFAP) Abs, and anti-serum-lymphocytoxic Abs (LCA) [50]. Systemic antibodies include antiphospholipid (aPL)/cardiolipin (aCL) Abs, lupus anticoagulant (LAC), anti-beta 2- glycoprotein I (2GPI) Abs, anti-ribosomal P Abs (anti-P), anti-Ro Abs, anti-Sm Abs, antiendothelial Abs (AECA), anti-serine proteinase (anti-PR3/C-ANCA) Abs, and anti-Nedd5 Abs [50].
4.1. Antiphospholipid Antibodies (β2-Glycoprotein 1, Cardiolipin Anticardiolipin (Anti-CL) and Lupus Anticoagulant (LA)
The aPL antibodies have an affinity to, and therefore target, anionic phospholipids, including β2GPI (rather than being against anionic phospholipids, which their name would suggest [51]) in the plasma membrane that regulates the blood clotting cascade [52]. The subsequent activation of procoagulants promotes thrombosis and cerebral infarction [53], and aPL antibodies have been identified for focal and diffuse NPSLE symptoms such as cognitive dysfunction [54], seizures [55], stroke, transient ischemic attack [56,57], movement disorders, chorea [58,59], and myelopathy [60].
4.2. Ribosomal P Protein (Anti-Ribosmal P Ab)
Anti-ribosomal P (anti-Rib-P) antibodies are specific serological markers observed in patients [61]. Anti-ribosomal-P antibodies are located at the carboxy-terminal end of the 60S subunit of ribosomes and target three phosphorylated proteins, P0, P1, and P2 [62]. Anti-ribosomal-P-antibodies are believed to breach the BBB, penetrate neuronal cells, and inhibit protein synthesis [63,64,65]. Antibodies against ribosomal-P proteins are associated with diffuse NPSLE, psychosis, and clinically significant depression in patients [66,67,68]. Their presence may be a risk factor for the development of NPSLE [69] and a predictor of psychosis in patients already diagnosed with NPSLE [70]. These antibodies may also be associated with complications of the peripheral nervous system [71]. In animal studies, depressive behavior was noted when anti-ribosomal P antibodies were introduced into the cerebral ventricles [72]. These antibodies cross-react with the neuronal surface P antigen on the membranes of neurons in the hippocampus and can manifest as clinical depression [73,74]. The anti-Rib-P antibody can also cross-react with NMDA receptors, resulting in psychosis [75], although the presence of anti-rib-P is not always associated with NPSLE manifestations [76]. Therefore, the clinical significance of anti-rib-P antibodies remains controversial.
4.3. Anti-Human N-Methyl-D-Aspartate Receptor Abs (Anti-NMDA)
The NMDA receptor is an ionotropic glutamate receptor in the CNS that is responsible for synaptic plasticity and memory [77]. The NR2A and NR2B subunits are found in the hippocampus, amygdala, and hypothalamus [78]. NMDA receptors are tetramers composed of NR1 subunits and two of the four NR2 (A–D) subunits [79]. Anti-NMDAR encephalitis is an autoimmune neurological condition associated with SLE; however, its pathophysiology is not fully understood [80,81]. Anti-NR2 antibodies cross-react with anti-double-stranded DNA antibodies [82]. Anti-NR2 antibodies can enter the CNS via intrathecal IgG synthesis or by breaching the BBB [83]. The severity of BBB damage plays a significant role in diffuse NPSLE syndromes, including the potential acute confusional state, because it allows large titers of anti-NR2 to enter the CNS [84].
In NPSLE patients, anti-NR2 antibodies pathologically bind to the extracellular domains of the NR2A and/or NR2B subunits of the NMDA receptor. These autoantibodies have a much higher sensitivity to the NR2A subunit, resulting in excessive activation of the NMDA receptor [85]. Pathological NMDA receptor activation in patients has been found to manifest as epilepsy, encephalitis, schizophrenia, mania, stroke, and cognitive impairment [78,86].
4.4. Microtubule-Associated Protein (Anti-MAP-2 Ab)
MAP-2 is a cytoskeletal protein expressed primarily in neuronal cells that is responsible for microtubule nucleation and stabilization, and regulates organelle transport protein kinases involved in signal transduction [87,88]. Anti-MAP-2 antibodies are associated with neuronal injury and death and are significantly elevated in the CSF of patients with NPSLE [89]. Anti-MAP-2 antibodies are associated with neuropsychiatric symptoms, such as psychosis, schizophrenia [90], bipolar disorder [91], major depression [92], seizures, neuropathy, and cerebritis [42].
4.5. U1 Ribonucleoprotein (Anti-U1RNP Ab)
Anti-UIRNP antibodies are observed in autoimmune conditions such as mixed connective tissue disease (MCTD), systemic sclerosis (SSc), and systemic lupus erythematosus (SLE) [93]. The small nuclear ribonucleoproteins (snRNP) are RNA–protein complexes found in abundance in the nucleus and are involved in the processing of pre-mRNA and other proteins comprising the spliceosome [94]. Anti-U1RNP antibodies react with one or more of the three proteins (70-kD, A, and C) that are specifically present in the U1 RNP complex to form U1 small nuclear ribonucleoprotein (snRNP) [95]. The snRNP is a target of autoreactive B and T cells in several rheumatic diseases, including SLE [96]. Anti-U1 RNP antibodies range from 3 to 69 percent in patients with SLE [97]. Anti-U1RNP Ab is associated with NPSLE manifestations such as anxiety, seizures, and CVD [98].
4.6. Structural Endothelial Proteins (AECA)
Endothelial cells (ECs) are found on the inner walls of blood vessels and form a layer of cells referred to as the endothelium. Endothelial cells have not been previously considered as components of the immune system. ECs are important for regulating blood pressure and play important roles in coagulation, fibrinolysis, angiogenesis, and immune cell activation via both physiological and pathological processes [99]. The modulation of endothelial cells via the adaptive and innate immune systems plays an integral role in autoimmune diseases, as endothelial cells promote chronic inflammation via angiogenesis, attracting immune cells, and antigen presentation [100]. Anti-endothelial cell antibodies (AECA) are a heterogeneous group of autoantibodies directed against structural endothelial proteins, along with antigens on endothelial cells [101]. The activation of ECs leads to increased leukocyte adhesion, the activation of coagulation, and vascular thrombosis in a dose-dependent manner [102]. The pathologic activation of ECs results in endothelial injury and an increased risk of complications, such as atherosclerosis and vascular thrombosis, which are the most common causes of premature mortality in patients with SLE [103].
4.7. Triosephophate Isomerase (Anti-TPI Ab)
Triosephosphate isomerase (TPI) is a glycolytic enzyme found in neuronal and red blood cells that are involved in the interconversion of dihydroxyacetone phosphate (DHAP) and glyceraldehyde-3-phosphate (G3P) [104]. Anti-TPI antibodies have been associated with NPSLE with a higher frequency of aseptic meningitis and elevated serum IgG levels in anti-TPI-positive NPSLE patients compared to anti-TPI negative NPSLE patients [105]. Anti-TPI antibodies likely breach the BBB via meningeal inflammation. Anti-TPI antibodies form immune complexes in the CSF and activate the classical complement system, contributing to the pathogenesis of NPSLE [106].
4.8. Glyceraldehyde-3-Phosphate Dehydrogenase Antibodies (Anti-GAPDH)
GAPDH is a glycolytic enzyme that also plays a role in cell membrane fusion, microtubule bundling, nuclear RNA export, and DNA replication and repair [107]. Anti-GAPDH antibodies have been associated with increased disease activity, increased inflammation, and increased intracranial pressure in NPSLE patients [108]. GAPDH antibodies have been found in at least 50% of NPSLE patients with schizophrenia and major depression and may be a future potential biomarker of NPSLE [109]. A significant positive relationship between the levels of anti-GAPDH antibodies in the serum and detrimental cognitive and mood conditions (such as schizophrenia and major depression) in patients with SLE has been reported. The levels of anti-GAPDH antibodies were found to be higher in SLE patients exhibiting psychotic symptoms compared to those without such symptoms [109]. This finding is further supported by a study conducted by Sun and collaborators, which discovered that the levels of anti-GAPDH antibodies in the serum were significantly increased in NPSLE patients and were correlated with increased SLEDAI-2K, ESR, IgG, and IgM [108].
Anti-GAPDH antibodies are used in the immunodetection of the protein encoded by the GAPDH gene. GAPDH is an enzyme that catalyzes the sixth step of glycolysis and serves to break down glucose for energy and carbon molecules. In addition to its role in metabolism, GAPDH is involved in the initial stages of apoptosis and the oxidative stress response where GAPDH is translocated to the nucleus. Such actions may reflect the role of GAPDH in DNA repair or as one nuclear carrier for apoptotic molecules. GAPDH has also been found to bind specifically to proteins implicated in the pathogenesis of a variety of neurodegenerative disorders [110,111].
4.9. Anti-Aquaporin Four Antibodies (NMO-IgG/AQP4-Ab)
Anti-AQP4 antibodies, also known as NMO-IgG, have been identified in patients with NPSLE [112]. These antibodies target the Aquaporin 4 (AQP4) water channel protein, which is predominantly found in the central nervous system. In one study, it was found that these antibodies were present in a patient with transverse myelitis, a rare but serious complication of SLE [1,112]. However, they were not detectable in NPSLE patients with other neurological manifestations. This suggests that testing for NMO-IgG/AQP4-Ab positivity should be considered in patients presenting with SLE and TM [1,112].
4.10. Anti-Endothelial Cell Antibodies (AECAb)
Anti-endothelial cell antibodies (AECAb) are autoantibodies that target endothelial cells, which are the cells that line the interior surfaces of blood vessels [36]. In the context of neuropsychiatric systemic lupus erythematosus (NPSLE), these antibodies have been associated with various pathogenic mechanisms [2,113]. AECAb has been implicated in inducing a proadhesive and proinflammatory endothelial phenotype through nuclear factor kappa B (NF-κB) activation, with the involvement of an autocrine loop of interleukin-1β secretion [36].
AECAb may contribute to the dysfunction of BBB, a layer of cells that prevents harmful substances in the blood from crossing into the brain. The presence of AECAb in NPSLE patients could potentially lead to cerebrovascular ischemia as a result of a generally prothrombotic state [36].
4.11. Anti-Ubiquitin Carboxyl Hydrolase L 1 Antibodies (Anti-UCH-L1 Ab)
Anti-UCH-L1 Abs have been studied as potential biomarkers for NPSLE. In particular, the autoantibody against the amino acids 58–69 of UCH-L1 (UCH58-69) has shown significant diagnostic power in distinguishing NPSLE patients from SLE patients without neuropsychiatric symptoms. The specificity and sensitivity of anti-UCH58-69 were found to be 92.3% and 37.5%, respectively. Increased serum levels of anti-UCH58-69 were associated with an increased disease severity, suggesting that this autoantibody could be a novel serum biomarker for the non-invasive diagnosis of NPSLE. This might be applicable for early screening and diagnosis of NPSLE. However, it is important to note that these findings are based on research studies, and further validation is needed before these antibodies can be used in clinical practice [39,114]. In a study conducted by Li et al. in 2019, it was found through a randomized controlled trial that antibodies against UCH-L1 could serve as a reliable biomarker in the cerebrospinal fluid (CSF) for diagnosing NPSLE. The study also found that the levels of UCH-L1 in the CSF could indicate the severity of NPSLE [115]. In a similar vein, a recent study on autoantibodies showed that the autoantibody targeting the amino acids 58–69 of UCH-L1 (UCH58-69) demonstrated a high level of specificity and diagnostic significance in differentiating NPSLE patients from SLE patients without neuropsychiatric symptoms. The study further revealed that the levels of anti-UCH58-69 in the serum were considerably higher in NPSLE patients compared to SLE patients without neuropsychiatric symptoms, and these levels were found to be associated with the severity of the disease [36]. Table 2 illustrates autoantibodies associated with NPSLE.
Table 2. Illustrating common NPSLE-associated symptoms in NPSLE.
| Autoantibody | Location Isolated | Associated NPSLE Symptoms | References |
|---|---|---|---|
| Phospholipid: β2-glycoprotein 1 and cardiolipin (aCL-Ab) |
Serum, CSF | CVD, seizures, chorea cognitive dysfunction, psychosis, depression, headache | [54,55,56,57,58] |
| Ribosomal P protein (anti-ribosmal P Ab) |
Serum, CSF | psychosis, depression, cognitive impairment | [66,67,68] |
| NMDA receptor (anti-NMDA) |
Serum, CSF | depression cognitive dysfunction | [84] |
| MAP-2 (anti-MAP-2 Ab) | Serum, CSF | seizures, chorea, sensory neuropathy, psychosis, headache) | [90,91,92] |
| U1 ribonucleoprotein (Anti-U1RNP Ab) |
Serum, CSF | Diffuse NPSLE symptoms | [98] |
| Structural endothelial proteins (AECA) | Serum | Psychosis, depression | [102,103] |
| Triosephosphate isomerase(anti-TPI Ab) | Serum, CSF | aseptic meningitis | [105] |
| GAPDH (anti-GAPDH Ab) | Serum | Involved in various in neurodegenerative disorders, increased intracranial pressure, cognitive dysfunction | [108,110,111] |
Autoantibodies against autoantigens were detected in the sera of NPSLE patients, as well as in the CSF. However, the authors did not compare the prevalence of these autoantibodies in the sera of NPSLE and non-NPSLE patients, so it is not clear if they are specific to NPSLE. Based on their results, the autoantibody that has the highest possibility of being a marker of NPSLE is anti-SS-A, because it showed a significantly higher positive rate in the CSF of NPSLE patients than in the CSF of non-NPSLE patients, and it was also related to neuropsychiatric syndromes of the central nervous system in SLE patients. The authors used a human proteome microarray to screen for autoantibodies in the cerebrospinal fluid (CSF) of patients with neuropsychiatric systemic lupus erythematosus (NPSLE), a subtype of SLE that affects the nervous system. They identified autoantigens that were specifically associated with NPSLE, and found that they were enriched for functions involved in neurological diseases. They also found 22 autoantigens that were shared by NPSLE and non-NPSLE patients, and found that they were enriched for functions involved in inflammatory responses. They validated some of the candidate autoantigens using a focused autoantigen microarray and western blot, and confirmed that anti-SS-A and anti-PCNA autoantibodies were significantly associated with NPSLE. They also found that the titers of anti-RPLP2 and anti-SS-A autoantibodies in CSF and serum specimens were significantly correlated, suggesting that they leaked from the blood due to the compromised BBB [116].
This entry is adapted from the peer-reviewed paper 10.3390/molecules29040747
This entry is offline, you can click here to edit this entry!