Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Nonalcoholic fatty liver disease, recently re-named metabolic dysfunction-associated steatotic fatty liver disease, is considered the most prevalent liver disease worldwide. Its molecular initiation events are multiple and not always well-defined, comprising insulin resistance, chronic low-grade inflammation, gut dysbiosis, and mitochondrial dysfunction, all of them acting on genetic and epigenetic grounds.
- antibiotics
- use/misuse
- food residues
1. Introduction
NAFDL, recently re-named metabolic dysfunction-associated steatotic fatty liver disease (MASLD), even though the differences between the old and new acronym are negligible, is considered the most prevalent liver disease all over the world. We use the term NAFLD instead of MASLD throughout this text in light of the fact that the previous literature sources which we refer to use the old acronym [1]. What is more, the diagnostic terms “fatty liver”, “NAFLD”, and “NASH” were not associated with discomfort for the majority of patients. Although 26% of patients reported stigma related to overweight/obesity, only 8% disclosed a history of stigmatisation or discrimination due to NAFLD [2].
The risk factors of NAFLD are multiple and not always clearly defined, ranging from metabolic alterations (dyslipidaemia) to genetic/epigenetic traits [3], lifestyle modifications, including smoking [4], and even viral aetiology [5]. Obesity, mainly abdominal obesity, is one of the most important drivers [6]. Furthermore, patients with the severe form of NAFLD, i.e., nonalcoholic steatohepatitis (NASH) and obesity showed worsening of fibrosis versus those without obesity [7]. Still, sarcopenia is associated with increased risks of NAFLD and advanced fibrosis, independent of obesity or metabolic control [8]. Obviously, type 2 diabetes mellitus (T2DM) plays a determinant role, even though most clinical investigators suggest that NAFLD appearing first leads ultimately to the development of T2DM [9].
The mechanisms underlying NAFLD are even more complicated, including gene variants such as patatin-like phospholipase domain-containing protein 3, transmembrane 6 superfamily member 2, hydroxysteroid 17-beta dehydrogenase 13, membrane-bound O-acyltransferase domain-containing 7, and glucokinase regulator [10]. A key role is played by dysfunctional adipocytes, with the involvement of the mesenteric adipose tissue, adipokines such as adiponectin, the food intake hormone (leptin) and leptin resistance, as well as of another adipose tissue’s hormone, i.e., resistin [11]. It has been ascertained that insulin resistance (IR) is central to the onset and progression of NAFLD. Both innate and recruited immune cells, such as macrophages and T-cells, mediate the development of IR. Infiltrated macrophages in obese adipose tissue undergo a phenotypic switch from alternative M2 macrophages to classical M1 macrophages. Consequently, modifying the polarisation of resident and recruited macrophage/Kupffer cells is expected to pave the way to new therapeutic approaches in NAFLD [12]. Lipotoxicity is an important downstream signalling event. Free fatty acids (FFAs) activate apoptosis, including the up-regulation and increased number of death receptors such as Fas and TRAIL receptor 5, at the level of the plasma membrane, lysosomal permeabilisation, and endoplasmic reticulum stress, both coupled to mitochondrial dysfunction and in turn activating the mitochondrial pathway of apoptosis. Furthermore, FFAs set TLR4 signalling in motion, resulting in the up-regulation of several pro-inflammatory cytokines. Finally, other lipids such as free cholesterol and ceramide induce mitochondrial dysfunction and initiate the mitochondrial pathway of apoptosis [13]. Chronic low-grade inflammation represents a further mechanistic level of NAFLD. Inflammatory mediators that are biosynthesised in the liver and increased in NAFLD patients include C-reactive protein, interleukin (IL)-6, fibrinogen, and plasminogen activator inhibitor-1. Recent data lend credence to the fact that hepatic steatosis triggers IκB kinase-beta and nuclear factor-κB. Among the inducible transcription factors that control inflammatory gene expression, nuclear factor-κB plays a central and evolutionarily conserved role in coordinating the expression of various soluble pro-inflammatory mediators (cytokines and chemokines) and leukocyte adhesion molecules, as reviewed in [14]. Recent clues suggest that endoplasmic reticulum stress is involved in the development of lipid droplets and subsequent generation of reactive oxygen species (ROS) in the progression to NASH. The molecular processes caused by the disruption of endoplasmic reticulum homoeostasis, described as unfolded protein response, are associated with membrane biosynthesis, insulin action, inflammation, and apoptosis [15]. Gut microbiota dysbiosis significantly contributes to the pathogenesis and severity of NAFLD. Actually, relatively higher abundances of the genera Fusobacteria, and lower abundances of Oscillospira and Ruminococcus of Ruminococcaceae and Coprococcus of Lachnospiraceae, have been found in NAFLD patients, as well as other bacterial species such as Proteobacteria, Escherichia, and Enterobacteria. Also, Bacteroides were shown to be more frequent in patients with NASH. Another study showed the lower diversity of microbiota in the faeces of children with NAFLD and an increased number of Prevotellacopri. Finally, a low alpha bacterial diversity was linked with severe liver fibrosis, as reviewed in [16]. Other pathogenic factors and pathological mechanisms known to be involved in the complex disease progression of NAFLD involve programmed cell death include (j) autophagy of fat, referred to as lipophagy, that when impaired can lead to fat accumulation mediated by osteopontin, reduced levels of glycine N-methyltransferase, and phosphorylation of Jumonji-D3; (jj) ferroptosis, characterized by cytological changes that include reduced cell volume and increased mitochondrial membrane density, identified by iron dependence and lipid peroxidation; (jjj) apoptosis/necroptosis, whose key molecules include mixed lineage kinase domain-like and the receptor-interacting protein (RIP) protein kinase family members RIPK1 and RIPK3, with tumour necrosis factor receptor 1-mediated signal transduction being an example of the conversion between apoptosis and necroptosis; and (jjjj) pyroptosis, which may occur through the classic caspase 1-dependent pathway and the nonclassical caspase 4/5 (mouse caspase-11) pathway. All the above pathways are detailed in [17]. Mitochondrial dysfunction is a further molecular process leading to NAFLD due to the increased flux of FFAs in hepatocytes, which leads to increased mitochondrial fatty acid import and oxidation. Consequently, the overproduction of ROS damages mitochondrial membranes and may result in mitochondrial permeability transition pore formation with the consequent release of mtDNA in cytoplasm, which acts as a danger-associated molecular pattern and may activate the NLR family pyrin domain-containing 3 inflammasome with consequent maturation of the cytokine IL-1 beta and perpetuation of inflammation, with this latter mechanism being common to alcoholic liver disease [18]. Bile acids (BAs), beyond FFAs, hormone receptors and drugs, bind to nuclear receptors (NRs) and modulate their transcriptional activity. In hepatocytes and other cell types in the liver, NRs control multiple metabolic and inflammatory processes that influence the development of NAFLD/NASH [19]. It is essential to evidence that adiponectin is inversely linked to IR [20]. Glucocorticoids bear the potential to drive NAFLD, acting on both liver and adipose tissue. In their fasting state, they are able to mobilise lipids, increasing fatty acid delivery, and in their fed state, they can promote lipid accumulation [21]. Recent studies suggest that telomere shortening can lead to cellular dysfunction by impacting metabolism via a p53-dependent repression of mitochondrial biogenesis and function that results in dysfunctional mitochondria [22]. With NAFLD being more prevalent in the elderly [23], telomere length plays an important role in this very common liver disease in the sense that low length accounts for high NAFLD risk, making it plain that telomere shortening causes senescent cells to accumulate [24], Figure 1.
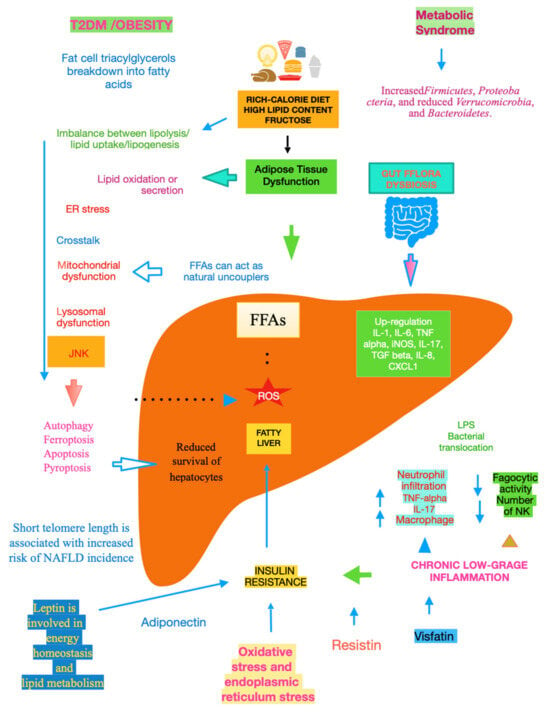
Figure 1. Main mechanisms of NAFLD.
2. The Connection between Antibiotics and Pathomechanisms of NAFLD
2.1. The Link between Antibiotics and NAFLD
In a nationwide case–control study, 2584 Swedish adults with histologically proven NAFLD, of which 56.6% had simple steatosis, 14.8% had steatohepatitis, and 26.8% had non-cirrhotic fibrosis, diagnosed from January 2007 to April 2017, were included and matched to ≤5 12,646 controls for age, sex, calendar year, and county of residence. Antibiotic use was found to be a risk factor for incident NAFLD, especially in individuals without metabolic syndrome. The risk was very significant for fluoroquinolones and remained robust in sibling comparisons with whom individuals shared genetic and early environmental susceptibilities [60].
Concerning the likely mechanisms, very recent data show that intestinal barrier dysfunction in diet-induced NAFLD in male C57BL/6J mice, either pair-fed a liquid control diet or fat- and fructose-rich diet +/− antibiotics (ampicillin/vancomycin/metronidazole/gentamycin) for seven weeks were not based on changes in intestinal microbiota but rather on altered intestinal nitric oxide (NO) homeostasis induced by fructose [61]. It has been shown that the development of NAFLD with starting inflammation was linked to impaired intestinal barrier function, increased NO levels, and a loss of arginase activity in the small intestines of female C57BL/6J mice that were pair-fed with a liquid control diet or a fat-, fructose-, and cholesterol-rich diet for eight weeks [62]. Treating mice with a variety of antibiotics (cefoperazone, clindamycin, and vancomycin) to create distinct microbial environments in the large intestine led to a significant loss of secondary BAs, such as deoxycholate, lithocholate, ursodeoxycholate, hyodeoxycholate, and ω-muricholate. These changes were related to the loss of specific microbiota community members, such as the Lachnospiraceae and Ruminococcaceae families [63]. Compared to controls, mice with the progressive form of NAFLD, i.e., NASH, had lower concentrations of secondary BAs in their portal blood and bile, while systemic BA concentrations were not significantly altered [64].
2.2. Other Molecular Initiation Events
It is not surprising that antibiotics act not only on the bacteria of exogenous infections, but also on the mitochondria from the human microbiome, explaining why their use bears various adverse side effects, such as mitochondrial dysfunction [65], increased ROS generation, and decreased ATP production with increased folding of the inner membrane, all of these mechanisms having a role in obesity and T2DM [66]. More and more data indicate that hepatic mitochondrial dysfunction is crucial to the pathogenesis of NAFLD [67]. We have previously underlined that low telomere length accounts for high NAFLD risk. Surprisingly, interesting findings showed that ofloxacin and levofloxacin, quinolones that are largely prescribed, were characterised at low concentrations by decreased telomerase activity, evaluated by absorbance values that were lower than the control cells, indicating a reduction in the rate of cell proliferation [68]. However, it should be stressed that azithromycin in turn attacks senescent cells, efficiently removing almost the totality of them, thus acting as a potent senolytic agent [69]. Gut microbiota influence host physiology by epigenetic regulation, ending up in chemical donors for DNA or histone modifications, or modifying enzyme expression and/or activity, or generating host-cell-intrinsic processes that direct epigenetic pathways. SCFAs represent another important group of epigenetically relevant molecules that are exclusively produced by commensal microbes through the fermentation of complex non-digestible carbohydrates and fibre [70]. It is well known that higher SCFA excretion was associated with evidence of gut dysbiosis, gut permeability, and excess adiposity [71]. It is noteworthy that serum concentrations of isobutyrate and methylbutyrate and their involvement in glucose metabolism, and in regulating insulin sensitivity and lipogenesis through diverse pathways, were significantly and negatively correlated with NAFLD severity [72].
Circulating meal-associated leptin and ghrelin levels and BMI changed significantly after H. pylori eradication, providing direct evidence that H. pylori colonisation, but also antibiotics, have effects on body morphometry [73]. Novel results showed that gut dysbiosis due to long-term use of systemic antibiotics can impact oral microbiota and aggravate periodontitis. Furthermore, the expression of cytokines related to Th17 was increased, while transcription factors and cytokines related to Treg were decreased in the periodontal tissue [74]. There is a close association between NAFLD and periodontal disease, as confirmed by epidemiological studies, basic research, and immunology, with Th17 being a key molecule for explaining the relationship between these two diseases [75].
This entry is adapted from the peer-reviewed paper 10.3390/ijms25041993
This entry is offline, you can click here to edit this entry!