Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
There has been a widespread adoption of hypomethylating agents (HMA: 5-Azacytidine (5-Aza)/decitabine) and venetoclax (Ven) for the treatment of acute myeloid leukemia (AML); however, the mechanisms behind the combination’s synergy are poorly understood. Monotherapy often encounters resistance, leading to suboptimal outcomes; however, the combination of HMA and Ven has demonstrated substantial improvements in treatment responses. This study elucidates multiple synergistic pathways contributing to this enhanced therapeutic effect.
- Venetoclax-5-Azacytidine
- hypomethylating agents
- acute myeloid leukemia
- myeloid malignancies
1. Introduction
Acute myeloid leukemia (AML) is a heterogenous condition, generally affecting older individuals [1]. The ineligibility of such patients to intesive chemotherapy has led to the search for newer therapies in recent years. One such example of a newer combination of hypomethylating agents (HMAs) and a selective oral BCL-2 inhibitor (Venetoclax, Ven) has made significant strides and gained FDA approval in recent years [2].
HMAs, such as 5-azacytidine (Aza) and decitabine (Dec), function by promoting the blocking of DNA methyltransferase 1 (DNMT1). This process results in the demethylation of CpG islands [3] and an increase in the expression of genes necessary for normal myeloid hematopoiesis. HMAs, thus, play an integral role in the treatment of myelodysplastic neoplasms and acute myeloid leukemia (AML), when cellular differentiation blocks arrest maturation [4,5,6].
Normal cellular senescence via apoptosis, regulated by a balance between pro-apoptotic (Bax, Bak, Bad, Bid, Puma, Bim, Noxa) and anti-apoptotic proteins (Bcl-2, Bcl-xL, Bcl-w, Mcl-1), is lost among cancer cells, characterized by increased expression of anti-apoptotic protiens [7]. An anti-apoptotic protein prevents apoptosis by binding to and clearing pro-apoptotic proteins.
In 2014, Pan et al. [8] showed the efficacy of Ven (ABT-199), a potent, selective oral inhibitor of BCL-2, on multiple AML cell lines and patient-derived xenografts at nanomolar concentrations. Notably, the cytotoxic effects of Ven (Venetoclax) were predominantly unaffected by mutational status and were sustained in primary AML blast cells that were resistant to chemotherapy. In a phase II clinical trial, Konopleva et al. [9] showed evidence for the efficacy of Ven monotherapy (19% overall response rate), particularly among patients with IDH1/2 mutations.
2. Mechanisms of Resistance against Monotherapies with Ven or HMA in AML Cell Lines
2.1. Resistance against Ven
The details of Ven resistance have been described elsewhere [16]. Briefly, the primary mechanism involves the overexpression of anti-apoptotic proteins other than BCL-2, i.e., BCL2-A1, MCL-1, and BCL-xL, that sequester pro-apoptotic proteins (e.g., BIM, BAX) [17,18,19]. Broadly, these mechanisms include the inability to trigger apoptosis due to inactivation of BAX/BIM, TP53, and PMA1P1 (NOXA); decreased BCL2 expression, hence lesser activity of Ven (a selective BCL-2 inhibitor); and alterations in mitochondrial function and cellular metabolism [20].
Another resistance mechanism includes stimulation of signaling pathways by mutant KRAS/PTPN11 or FMS-like tyrosine kinase 3 (FLT3) proteins. These mutations could either be intrinsic or emerge secondary to Ven-based therapy, and they carry an adverse prognosis [21]. The anti-apoptotic factors MCL-1 and BCL2-A1 are upregulated due to a mutation in the KRAS gene, whereas a mutation in PTPN11 results in the upregulation of the anti-apoptotic factors MCL-1 and BCL-xL. KRAS mutations also downregulate BCL-2 and BAX [19]. Venetoclax resistance is seen in FLT3-ITD mutations due to upregulation of the anti-apoptotic factors BCL-xL and MCL-1 via complex downstream pathways [22,23,24].
2.2. Resistance against HMA
The resistance mechanisms against HMA (Aza or Dec) are related factors intrinsic to tumor cells and extrinsic factors related to immune cells or other cells in the bone marrow, as discussed elsewhere [4]. Tumor cell-intrinsic factors are related to HMA transport (via human concentrative or equilibrative nucleoside transporter 1; hCNT1 or hENT1) into cells, activation (via kinases; UCK or DCK), incorporation into nucleic acids, and DNMT inhibition and metabolism via cytidine deaminases (CDAs). Low levels of transporters and decreased expression of kinases, leading to decreased incorporation of Aza or Dec into nucleic acids, contribute to HMA resistance [26,27,28,29].
HMAs can induce major antioxidant pathways mediated by the NF-E2-related factor (Nrf2), thus contributing to self-resistance [30]. Cheng JX et al. also reported that leukemia cells resistant to 5-Aza exhibit increased active chromatin organization associated with one of the methylcytosine (RNA: m5C) methytransferases, namely, NSUN1. NSUN1 establishes interactions with BRD4 and RNA polymerase II, resulting in the formation of an active chromatin structure resistant to 5-Aza but responsive to the inhibition of BRD4 [31]. Extrinsic factors are commonly related to, but not limited to, the bone marrow microenvironment.
3. Ven–HMA Synergy Mechanisms
3.1. Preclinical Data
3.1.1. HMA-Mediated Downregulation of MCL-1 Levels
Tsao et al. [32] initially reported that co-administration of 5-Aza and ABT-737 (inhibitor of BCL-2 and Bcl-xL) could increase mitochondrial outer membrane permeability among AML cells. This was reflected via the activation of BAX protein and loss of the mitochondrial membrane potential. They noted that the 5-Aza and ABT-737 combination synergistically induced apoptosis independent of p53 expression in these cells, which was not the case when these agents were used as single agents. Elevated levels of anti-apoptotic MCL-1 are known for conferring resistance to ABT-737. The application of 5-AZA reduced the expression of MCL-1 in these cells, independent of p53, leading to enhanced AML cell apoptosis.
The role of MCL-1 inhibition (by S63845, a specific MCL-1 inhibitor) in synergizing venetoclax activity was further assessed by Hormi et al. [33]. They observed that despite relatively weaker expression of MCL-1 compared to BCL-2, S63845 prompted apoptosis in AML cells and exhibited a robust synergistic effect with Venetoclax.
3.1.2. BCL2 Family Protein as 5-Aza-Sensitizing Targets
Bogenberger et al. [34] noted greater sensitization to 5-Aza (three- to fourfold) in AML cells with RNAi-mediated knockdown of BCL-xL. Furthermore, they focused on the involvement of three prominent anti-apoptotic proteins—BCL-2, MCL-1, and BCL-XL—in influencing cell proliferation and responsiveness to 5-Aza. Notably, the siRNA-mediated silencing of BCL-XL or MCL-1 substantially diminished viability across the majority of the examined AML cell lines. In contrast, the impact of BCL-2 siRNA was comparatively less pronounced. The silencing of BCL-xL strongly sensitized particular types of erythroid cells, while MCL-1 sensitized more broadly across AML cells.
Navitoclax (ABT-263, its tool compound was ABT-737), a combined inhibitor of BCL-xL, BCL-w, and BCL2 and Venetoclax (ABT-199, selective BCL2 inhibitor) were utilized to assess the relative potency of these agents in sensitizing the AML cells for 5-Aza. Higher concentrations of ABT-199 compared to ABT-737 were required for the enhancement of Aza activity. The association was dose-dependent with ABT-737 but not with ABT-199, except for MDS-L cells. A greater synergy was noted with ABT-737 than ABT-199 in most cell lines. The sensitization was mediated by enhancing apoptosis; hence, they concluded that ABT-737 had greater potency when compared to ABT-199 and in combination with 5-Aza.
3.1.3. Combination Disrupts Energy Metabolism and Targets Leukemia Stem Cells (LSCs)
Extended exposure to azacitidine (oral; CC-486) harnesses sustained DNMT1 loss and sustained hypomethylation. This can lead to preferential targeting of immature LSCs, potentially addressing myeloid maturation arrests by facilitating differentiation [35]. Pollyea et al. [36] reported that the combination of Ven + 5-Aza altered the tricarboxylic acid cycle, as indicated by a decrease in α-ketoglutarate levels and an increase in succinate levels in LSCs. This suggested a targeted inhibition of electron transport chain complex II, specifically in LSCs. In vitro modeling further validated the inhibition of complex II through the diminished glutathionylation of succinate dehydrogenase.
Together, these data suggest that the azacitidine-mediated differentiation of LSCs coupled with Ven-mediated programmed cell death further synergizes each agent’s activity. While the Ven–HMA combination targets oxidative phosphorylation in LSCs, there may be an additional role of HMA-mediated LSC differentiation that augments durable responses in AML patients.
3.1.4. Reactive Oxygen Species (ROS)-Dependent Antileukemic Activity
Ven augments HMA by inhibiting Nrf2 antioxidant pathway activation, induced by HMA. This leads to oxidative killing of AML cells, via an indirect underlying mechanism. The NF-E2-related factor 2 (Nrf2) antioxidant pathway is the key cellular mechanism that prevents oxidative cell damage. In the presence of oxidative stress and the generation of ROS, the transcription factor Nrf2 dissociates from its cytoplasmic assembly with Kelch-like ECH-associated protein 1 (Keap-1). Under normal circumstances, Keap-1 facilitates the ubiquitination and degradation of Nrf2 [37,38]. Upon release, Nrf2 translocates to the nucleus, where it binds to the antioxidant response elements in the promoters of several genes, including heme oxygenase-1 (HO-1) and NADP-quinone oxidoreductase-1 (NQO-1). These genes play a role in neutralizing ROS [37,38].
3.1.5. HMA (5-Aza) Induced “Priming” of the AML Cells for Ven-Induced Apoptosis
Jin et al. [41] demonstrated a novel non-epigenetic mechanism of action for 5-Aza and its synergistic activity with Ven through the integrated stress response (ISR)-mediated induction of PMAIP1 transcripts. They reported that 5-Aza pretreated AML cells in vitro were more sensitive to Ven than DMSO pretreated cells. The number of cells with active caspase-3/-7 was higher immediately following treatment with combined 5-Aza and Ven exposure than with either agent alone. Molecular analysis revealed 5-Aza dose- and time- dependent increases in the pro-apoptotic protein factors NOXA (PMAIP1) and PUMA (BBC3) in cells obtained from AML patients. Yet, chronic 5-Aza therapy did not exert a substantial impact on gene expression or methylation. The promoter regions and transcripts of PMAIP1 and BBC3 genes remained unmethylated and unchanged. They postulated a role for the ISR pathway in Aza-induced priming. ATF4, the ISR pathway’s main effector, can promote apoptosis through transcriptional activation of proapoptotic factors (including PMAIP1 (NOXA) and BBC3 (PUMA)). When AML cells were exposed to the Ven–Aza combination, the levels of activating transcription factor 4 (ATF4) were significantly elevated within 24 h, indicating the activation of the ISR pathway by 5-Aza. These preclinical findings underscore the clinical importance of initiating both therapies concomitantly, preferably within 24 h of HMA initiation [41,42]. The relative impact of NOXA and PUMA was delineated by deleting their respective genes (PMAIP1 and BBC3). A reduction in the magnitude of cell death from the 5-Aza/Ven combination was observed in PMAIP1-deleted cells but not in BBC3-deleted cells. Therefore, they came to the conclusion that, whereas 5-Aza therapy induces both NOXA and PUMA, only NOXA is essential for sensitizing AML cells to 5-Aza/Ven-induced apoptosis [41].
3.1.6. Overexpression or HMA-Mediated Restoration of Caspase-3/GSDME Significantly Increases Ven-Induced Pyroptosis
Ven induces pyroptosis through the cleavage of the gasdermin (GSDM) protein, a process mediated by caspase-3. Cleaved GSDM forms membrane pores, leading to cytokine release and/or programed lytic cell death, called pyroptosis [45]. Recently, Ye et al., via in vivo and in vitro experiments, showed that GSDME, but not GSDMA, GSDMB, GSDMC, or GSDMD, was cleaved upon Ven treatment. GSDME is suppressed in AML cell lines through promoter methylation, and reduced GSDME expression is strongly correlated with an unfavorable prognosis. They showed that the overexpression of GSDME, achieved through the demethylation of the GSDME gene or the restoration of GSDME expression mediated by HMA, markedly enhanced Ven-induced pyroptosis in AML [46].
3.1.7. Ven Augments HMA via Inhibiting De Novo Pyrimidine Synthesis
Upregulated de novo pyrimidine synthesis is the major adaptive resistance mechanism against HMA (Aza and decitabine) in MDS/AML cells [47]. The mitochondrial membrane potential is required for the functioning of the mitochondrial enzyme DHODH, a key enzyme of the de novo pyrimidine synthesis in cells. Ven, by inhibiting BCL-2, depolarizes the mitochondrial membrane and, hence, inhibits the DHODH enzyme. At concentrations that do not induce apoptosis, both Ven and the DHODH inhibitor teriflunomide substantially reduce the levels of cytidine- and deoxycytidine triphosphate (CTP, dCTP) in AML cells. This contributes to the downregulation of de novo pyrimidine synthesis, hence overcoming resistance against hypomethylating agents (HMAs) [48,49].
3.2. Clinical Data: Molecular Predictors for Response to Ven–HMA Combination
The FDA approved Ven–HMA as a frontline treatment for older adults aged 75 years or above, or those with comorbidities that make them unsuitable candidates for intensive induction chemotherapy [2,50]. In a pivotal randomized clinical trial, there was a significant improvement in the median OS (14.7 months for Ven + HMA vs. 9.6 months for placebo + HMA) and complete response rates (37% vs. 18%, respectively) [10].
The analysis of data from a phase 1b/2 study involving 209 patients treated with Ven 400 mg or 600 mg in combination with either HMA or LDAC revealed a CR rate of 83.7% for pts with IDH1/IDH2 mutations, 84.6% for patients with NPM1 mutations, 59.5% for patients with TP53 mutations, and 53.3% for patients with FLT3 mutations [51]. In the pivotal VIALE-A trial [10] (n = 431, median age of 76-years), patients with IDH1 or IDH2 mutations at baseline had better OS at 12 months, with 5-Aza + Ven (66.8%), as compared to 35.7% with 5-Aza + placebo group (with the hazard ratio for death, 0.35; 95% CI, 0.20 to 0.60; p < 0.001). Superior responses were noted with Ven + HMA in patients with IDH1/2, FLT3, NPM1, and TP53 genes. In another study, DiNardo et al. reported high response rates and sustained remissions among patients with NPM1 or IDH2 mutations (2-year OS of 71.8% and 79.5%, respectively). The study identified that primary and adaptive resistance to Ven-based combinations was predominantly associated with mutations in FLT3, RAS, or TP53 genes [52].
Morisa et al. retrospectively performed analysis of 86 patients (44 newly diagnosed with AML and 42 with relapsed/refractory AML) who received an HMA–Ven combination. In the upfront setting, logistic regression analyses revealed that the presence of the CEPBA mutation conferred improved CR/CRi rates. Each of the four patients with CEPBA mutation attained CR, in contrast to 51% (18 of 35) of CEPBA wild-type patients. The median OS for patients receiving upfront HMA–Ven was 11 months (95% CI; 8–23 months). It was higher among those recieving CR compared to those not achieving complete response (17 months vs. 3 months). For relapsed/refractory disease, the presence of JAK2 (p = 0.03), DNMT3A (p < 0.01), and BCOR (p = 0.04) mutations was identified as predictors of CR/CRi. Median overall survival for relapsed/refractory AML patients was 5 months (95% CI, 3–9 months), which was higher among those patients achieving complete response (15 months vs. 3 months) vs. those not achieving CR/CRi [12].
.3. Superior Response with Ven–HMA Compared to Ven-LDAC
Clinical studies have reported superior response with Ven–HMA compared with Ven low-dose cytarabine (LDAC) therapy in newly diagnosed (CCR rate: 66.4% (Ven–HMA in VIAL-A trial) vs. 48% (LDAC-Ven in VIAL-C trial)) and r/r AML patients (CCR rate of 37% vs. 11%, respectively, in retrospective study) [10,11,57].
The precise mechanism of this difference in response rates is yet to be explored and likely extends beyond the different study populations in the two landmark studies that led to the FDA approval of both regimens. It relates to the mechanism of action or potential drug–drug interactions, patient characteristics, and specific genetic mutations, all of which can significantly influence response rates. HMA and Ven synergize together via multiple mechanisms (Figure 1). However, LDAC, also known as, arabinosylcytosine (ARA-C), undergoes conversion into its triphosphate form within the cell. This form competes with cytidine to integrate into the DNA. The sugar moiety of cytarabine impedes the rotation of the molecule within the DNA, leading to the cessation of DNA replication, particularly during the S phase of the cell cycle. Additionally, DNA replication and repair are halted due to the inhibition of DNA polymerase by cytarabine [58]. Variation in single nucleotide polymorphisms (SNPs) within the genes responsible for the transport, activation, and inactivation of cytarabine can impact the intracellular ara-CTP levels. These SNPs can influence the expression and activity of these genes and, consequently, can affect the clinical outcome of patients treated with ara-C [59].
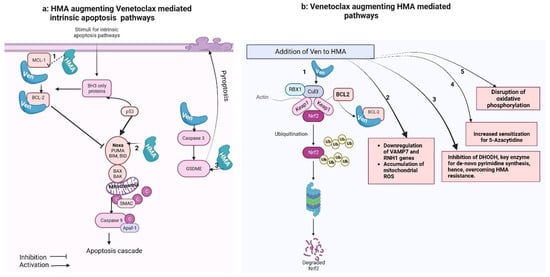
Figure 1. Venetoclax–hypomethylating synergy mechanisms. These figures were made using Biorender website (Biorender.com).
Figure 1a shows that the intrinsic apoptotic pathway is regulated by proapoptotic (BAX, BAK, BID, BAD, BIM, PUMA) and anti-apoptotic (BCL-2, MCL-1, BCL-xL) factors. Pro-apoptotic activator proteins, once triggered by a stimulus, activate the central effectors of apoptosis (BAX and BAK) that undergo confirmational change and form pores in the mitochondrial membrane, causing miMOMP (minority-mitochondrial outer membrane permeabilization) and release of cytochrome-C. Free cytochrome-C then binds with Apaf-1 (apoptosome), which activates caspase-9 and triggers the apoptotic cascade. Anti-apoptotic proteins, when released, bind with and neutralize anti-apoptotic proteins.
Figure 1b shows that Venetoclax synergizes with HMA-mediated cancer cell killing: (1) The primary cellular mechanism for averting oxidative cell damage is the NF-E2-related factor 2 (Nrf2) antioxidant pathway. Exposure to oxidative stress and reactive oxygen species (ROS) leads to dissociation of the transcription factor Nrf2 from its cytoplasmic adaptor Kelch-like ECH-associated protein 1 (Keap-1). In the absence of oxidative stress, Keap-1 mediates the ubiquitination and degradation of Nrf2. Once released, Nrf2 translocate to the nucleus and activates genes, which mediate neutralization of ROS (Baird and Dinkova-Kostova, 2011 [38]; Nguyen et al., 2009 [37]). This pathway is also induced by HMA.
This entry is adapted from the peer-reviewed paper 10.3390/ijms25010484
This entry is offline, you can click here to edit this entry!