Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
The presence and impact of toxins have been detected in various regions worldwide ever since the discovery of azaspiracids (AZAs) in 1995. These toxins have had detrimental effects on marine resource utilization, marine environmental protection, and fishery production. Over the course of more than two decades of research and development, scientists from all over the world have conducted comprehensive studies on the in vivo metabolism, in vitro synthesis methods, pathogenic mechanisms, and toxicology of these toxins.
- azaspiracids
- detection
- biosynthesis
1. Introduction
Since the 20th century, human beings have progressively explored and exploited marine resources. The high nutritional value of shellfish, along with their abundance of unsaturated fatty acids, has been a significant factor in attracting human consumption. However, shellfish possess self-protective mechanisms that often lead to the production of toxins and other harmful substances for humans. Additionally, as apex predators in the planktonic food chain, shellfish can accumulate toxins produced by algae [1].
Azaspiracids (AZAs) are a type of polyether toxin (Figure 1) that was initially discovered in an episode of food poisoning in Ireland in 1995 [2]. Several individuals in Ireland exhibited symptoms of diarrhea after consuming mussels, leading to the isolation and identification of these toxins [3]. AZAs were originally named Killary Toxin or KT-3 [4], and their name was later changed to Azaspiracids to better reflect the chemical structural formula. Azaspiracids are known to be produced by the Protists Azadinium [5,6] and Amphidoma [7,8], which belong to the order Lumbar Flagellate. Furthermore, these toxins have been reported and detected in various shellfish species, such as oysters, scallops, and clams [9,10,11].
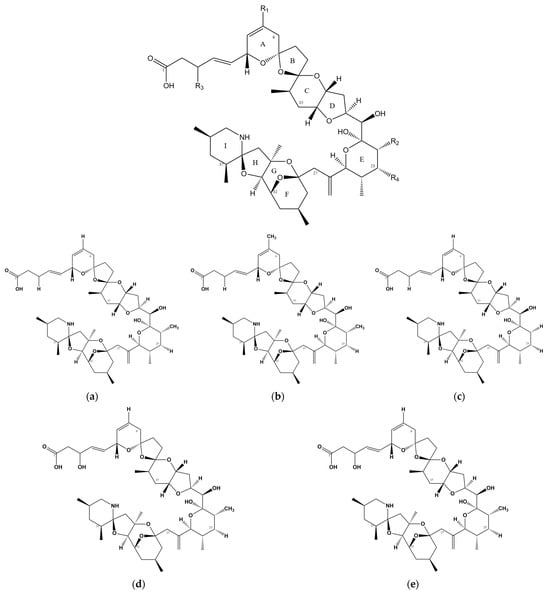
Figure 1. Chemical structure of AZAs. (a) Chemical structure of AZA-1, (b) chemical structure of AZA-2, (c) chemical structure of AZA-3, (d) chemical structure of AZA-6, and (e) chemical structure of AZA-7.
2. Toxin Distribution
Currently, there are reports of cases of AZA poisoning on many continents, including North America [15], South America [16], Africa [17], Europe [18], and Asia [19].(Figure 2).
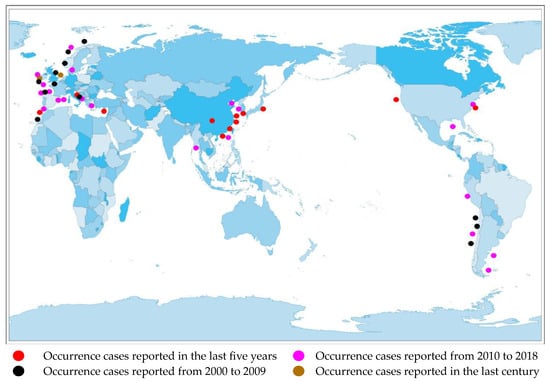
Figure 2. Distribution of AZA separation or occurrence.
3. Toxicology and Pathology
The acute lethal efficacy LD50 values of oral AZA-1, AZA-2, and AZA-3 in mice were determined to be 443 μg/kg, 626 μg/kg, and 875 μg/kg [20], respectively. The final symptoms of oral administration of AZAs in mice include immobility, sternal lateral immobility, tremors, abdominal breathing, hypothermia, and cyanosis. Although diarrhea is the main toxic sign of human ingestion of seafood contaminated with AZAs, oral exposure from AZA-1 to AZA-3 in mice did not cause significant diarrhea. The lethal dose of intraperitoneal injection in mice targeting AZA-1, AZA-2, and AZA-3 is 200 μg/kg, 110 μg/kg, and 140 μg/kg [12]. The signs and symptoms observed after intraperitoneal injection of purified AZAs in mice include progressive paralysis of the limbs, difficulty breathing, and pre-death convulsions [21]. Regarding the determination of 8 AZA-1 analogs and 12 fragments from the synthesis process of AZA-1, it was found that they have very low or almost no toxic effects compared to AZA-1 itself, indicating that the entire AZA-1 molecule and its stereo-orientation are necessary for exerting toxic effects. Animals subjected to AZA treatment exhibit organ swelling, along with the presence of fat droplets and vacuoles in liver cells [22]. The villi of the small intestine become blunt, accompanied by a reduction in the thickness of the brush-like edge. Additionally, there is a mild to moderate increase in apoptotic cells and infiltrating multi-nucleated cells in the mucosal lamina propria. Depletion of white medullary lymph nodes and lymphocyte necrosis are observed in the spleen. Hepatocellular necrosis is evident in liver cells. Prolonged exposure to small amounts of AZA toxins can lead to a decrease in digestive epithelial cells, increased lipid consumption, and an accumulation of lipofuscin [23]. The data on the oral toxicity of toxins in combination with other toxins indicate that neither the combination with OA (okadaic acid) nor YTX (yessotoxin) showed an increase in toxicity, and no overlapping or synergistic effects were found, only gastrointestinal symptoms were observed [24,25].
There are currently no reports on the long-term effects of AZA toxins on humans, but according to the European Food Safety Authority (EFSA), the lowest observed adverse effect level for individuals with a body weight of 60 kg is 1.9 μg AZA-1 equivalents/kg, and based on this, the most acute reference dose is calculated to be 0.2 μg AZA-1 equivalents/kg body weight [26].
4. Toxic Mechanism
Currently, there is a lack of definitive experimental research regarding the therapeutic mechanism of AZAs that would indicate its specific target-blocking properties. However, several studies have demonstrated its ability to influence cell electrical activity by affecting potassium ion channels [27], sodium ion channels [28], chloride ion channels [29], and calcium ion channels [30] (Figure 3). However, the specific mode of action, whether through interactions or intermediates, remains to be further explored. Specifically, its effect on the sodium ion channel is limited to the modulation of a fast sodium channel flow rate, without inducing channel inactivation. However, under the action of high-concentration toxins (200 nM), the proportion of channel inhibition can reach 60%, seriously affecting the process of cell depolarization [28]. AZAs can also inhibit sodium current through Nav 1.6 channels in the presence of glutamic acid [31], indicating that AZA poisoning may be due to its synergistic effect on some metabolites. Regarding the mechanism of potassium ion channels, it has been reported that AZAs can inhibit hERG channels (hERG channels play a very important role in myocardial repolarization) [27], and experimental data have shown that it can affect the quantity of hERG in the membrane in rats [32]. Rats treated with different doses of toxins (11 or 55 μg/kg) experienced partial PR interval prolongation and heart rate changes due to potassium ion channel blockade [32]. Inhibition of Ca2+ channels primarily affects the storage channels responsible for intracellular calcium ions, thereby giving rise to neurological symptoms [30].
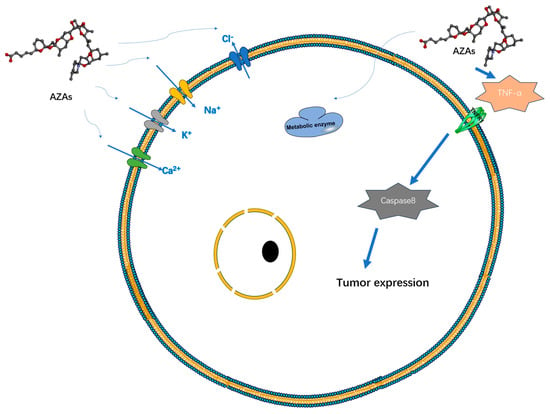
Figure 3. Possible targets and modes of action of AZAs currently reported.
Additionally, experimental data suggest that AZA-1 may act as a tumor initiator [33]. The induction mechanism here may be related to the abundant production of TNF-α and can induce the expression of early response genes jun B, jun D, c-fos, c-jun, fos B, and fra-1 to achieve tumor induction and occurrence. Repeated administration of AZA in mice led to a significant increase in lung tumors, as well as inducing lymph necrosis in tissues such as the small intestine, spleen, and thymus [34]. Prolonged exposure to AZAs in the environment can result in alterations in cellular cytoskeletons and reduced metabolic activity in human cells [28]. Certain experiments have demonstrated an upregulation of mRNA expression of genes associated with cholesterol synthesis and glycolysis following treatment with AZA toxins, suggesting a potential mechanism for AZAs in modulating cellular metabolism [35].
The gastrointestinal symptoms associated with AZA exposure may arise from alterations in the human intestinal glial system which can impact the integrity of the intestinal barrier. These changes include, but are not limited to, induced neuronal alterations, oxidative stress, disruption of the cell cycle, and an increase in specific enteric glial cell (EGC) markers [36]. Furthermore, the synergistic effects of multiple toxins present in the natural marine environment can enhance the virulence of AZAs [37]. Additionally, AZA-1 has been found to have a partial promoting effect on cell apoptosis and induce an increase in genetic toxicity. In terms of the blood system, AZAs can affect the damage to immune system cell lysosomes, consequently impacting phagocytic function [23].
Heart cells subjected to AZA-1 treatment exhibited heightened levels of apoptotic markers, including caspase-3 and -8, cleavage of PARP, and upregulation of Fas ligands [38]. These molecular changes are reflected at the tissue level, resulting in alterations in arterial blood pressure and deposition of cardiac collagen. Long-term experimental studies have demonstrated that AZAs can induce structural changes in the heart that contribute to heart failure [39] and provoke arrhythmias by modulating ion channels [32] (Figure 3).
In summary, although the activity of toxins as inhibitors of PP [40], kinases, and GPCR or as inhibitors of actin polymerization/depolymerization has been experimentally overturned [41], other experiments have shown their cytotoxicity, affecting cytoskeleton arrangement, promoting tumors, and potentially affecting the activity of multiple ion channels. However, at this point in time, there seems to be no scientific consensus on a specific target or mechanism of AZAs that can jointly explain the various effects observed in experiments and the gastrointestinal symptoms observed in exposed individuals, so further exploration of treatment is needed in the future.
5. AZA Analogs from Different Sources
The initial characterization of AZA and its analogs was conducted in 1998 using mass spectrometry (MS) and nuclear magnetic resonance (NMR) techniques [3]. Over 60 types of AZA analogs have been identified, with the majority being produced through metabolic processes in mussels.
AZA-38 and 39 are primarily produced by Amphidoma languida, a small dinoflagellate species belonging to the Amphidomataceae family. Recently, these toxins have undergone structural modifications [42]. The toxins produced by different ribosomal subtypes of Azadinium spinosum exhibit variations, with subtype A mainly producing AZA-1 and AZA-2, while subtype B primarily produces AZA-11 and AZA-51 [43]. In the case of ribosomal subtype A of Azadinium pomorum, there is either no production or only a minimal amount of AZAs, whereas ribosomal subtype C can generate AZA-40 and AZA-2 [44]. Different strains of Azadinium pomorum produce the hydroxylation product AZA-42 from AZA-41 and the dehydrogenation product AZA-62 from AZA-11. The former strain is isolated from the South China Sea, while the latter strain is isolated from the northern coast of Chile [45]. Furthermore, AZA-59 is the sole AZA toxin produced by Azadinium pomorum strains isolated from the Pacific northwest coast of the United States.
6. Synthesis In Vivo
To date, a majority of studies have pointed to the blue mussel, Mytilus edulis, as the primary vector for AZAs. However, other organisms such as mollusks, arthropods, and echinoderms have also been reported as potential vectors [46]. In mussels, several AZAs undergo acyl ester formation, with some studies suggesting that these esters exhibit higher toxicity than the free toxins themselves [47]. The average distribution of AZAs in mussels is as follows: hepatopancreas (60.6%), gills (12.0%), and adductor muscle (27.4%) [48].
AZA has been detected in various bivalve mollusks, including oysters (Crassostrea gigas), scallops (Pecten maximus), clams (Tapes filipinarium), and cockles (Cardium rule), as well as in numerous phytoplankton species [10]. In the experiment involving feeding blue mussels (Mytilus edulis) with toxins, it was found that AZA-17 and AZA-19 were mainly fast metabolites feeding AZA-1 and AZA-2 [49], indicating that carboxylation of methyl groups at the C22 position is a dominant metabolic pathway, while hydroxylation and decarboxylation are secondary degradation pathways. Notably, AZA-65 and AZA-66 have been identified as intermediate products in the conversion of AZA-1 and AZA-2 to AZA-17 and AZA-19. In mussels, the expression of AZA-1-3 can lead to the production of AZA-8, AZA-12, and AZA-5 through C-23 α hydroxylation. Additionally, the double hydroxylation of AZA-67 and AZA-68 can generate AZA-1 and AZA-2 as secondary metabolites [14].
Experimental evidence has demonstrated a correlation between toxin production in algae and temperature, with the highest toxin concentrations observed at 26 °C [50].
AZA-1 treated with rat liver microsomal extract undergoes oxidation at the F ring and can bind with glucuronic acid at C1 to generate glucuronides [51].
7. Synthesis In Vitro
In the synthesis of AZAs, the FGHI ring exhibits relatively high stability in the region spanning from C-26 to C-40, whereas the variability is primarily observed in the C-1 to C-22 [52] region. As early as 2008, the structure of the AZA-1 compound was fragmented and partially synthesized, encompassing the synthesis of the E ring, HI ring, CD ring, and FG ring. The conversion of furan into the ABCD ring can be achieved through a single oxygen-initiated one-pot process [53]. Subsequently, the combination of the EFGHI ring and the ABCD ring enables the complete synthesis of AZA-1 [54]. The artificial in vitro synthesis of fragments from C-22 to C-40 has been enhanced and refined [55]. Currently, through the full synthesis of AZA-1 and continuous modification and optimization of reaction conditions, an artificial stereoisomeric composite closely resembling the structure of natural AZA-3 has been achieved [56].
This entry is adapted from the peer-reviewed paper 10.3390/md22020079
This entry is offline, you can click here to edit this entry!