Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Gastroenterology & Hepatology
Ecological theories suggest that environmental factors significantly influence obesity risk and related syndemic morbidities, including metabolically abnormal obesity associated with nonalcoholic fatty liver disease (MASLD). These factors encompass anthropogenic influences and endocrine-disrupting chemicals (EDCs), synergistically interacting to induce metabolic discrepancies, notably in early life, and disrupt metabolic processes in adulthood.
- MASLD
- EDCs
- insulin-resistance
- children
1. Environmental EDCs and Sources of Exposure
EDCs encompass a broad spectrum of both natural and human-produced substances, originating from persistent organic pollutants (such as dioxins, benzo[a]pyrene and polychlorinated biphenyls (PCBs)), organochlorines (dichlorodiphenyltrichloroethane (DDT)), plasticizers (bisphenol A (BPA)), phthalates (di-2-ethylhexyl phthalate (DEHP)), organotins (tributyltin (polyfluoroalkyls and perfluorooctane sulfonate (PFOS))) and other pesticides (cypermethrin (CYP), atrazine (ATZ) and carbendazim). Approximately 1000 chemicals have been identified that meet the criteria for being classified as EDCs. These compounds are used across a diverse array of consumer products, including but not limited to food packaging, building materials, pesticides, clothing, detergents, plastics, and medical equipment. Some industrial process-related chemicals lead to unintentional contamination of food, water, and air. This exposes individuals through oral ingestion, dermal contact, inhalation, and even subcutaneous and intravenous routes (via medical equipment) [5].
The Stockholm Convention has identified a cluster of compounds termed the “dirty dozen”, which encompass pesticides, industrial chemicals, and their byproducts. This list includes aldrin, chlordane, toxaphene, mirex, hexachlorobenzene, heptachlor, furans, endrins, polychlorinated biphenyls (PCBs) and bisphenyl polybrominated (PBBs), polyfluoroalkyl substances (PFOS), dioxins, plasticizers (BPA, phthalates) pesticides (i.e., dichlorodiphenyltrichloroethane (DDT)), diethylstilbestrol (DES), and heavy metals (arsenic, cadmium and mercury) [26] (Table 3). Polycyclic aromatic hydrocarbons (PAHs) are generated through open combustion, natural filtration of oil or coal deposits, and incomplete combustion of various materials like coal, oil, gas, wood, waste, and tobacco [26]. PAHs can contaminate the environment by binding to airborne particles or adhering to foods cooked or roasted at high temperatures. Fluorinated chemicals (PFCs), extensively used in the manufacturing of cookware, clothing, food packaging, and firefighting materials, are widespread pollutants found in household environments. Due to their slow elimination from the body, they are classified as persistent organic pollutants (POPs), with a half-life ranging from approximately 2 to 8 years [26,27]. Bisphenol A (BPA) stands out as a notable EDC and is ranked third among the most significant pollutants according to the Environmental Protection Agency (EPA). BPA is an organic compound characterized by 2-phenolic rings connected by a methyl bridge with 2-methyl functional groups, classified in the phenol group. Due to its high strength as well as its hardness and transparency, BPA is widely used in industry as a material with which to produce phenolic resins, polyacrylates and polyesters and, above all, to produce plastics used in the manufacturing of everyday products [28]. Exposure to BPA happens via pollution, inhalation, and skin contact, but the most important route is from the consumption of contaminated foods, canned products, pre-packaged foods, packaged infant formula, baby bottles and containers used for food storage [26,27,28,29] (Table 3).
Table 3. Main EDCs implicated in hepatic injury.
| EDCs | Name | Molecular Targets | Study Model |
|---|---|---|---|
| Solvents/lubricants | Bisphenyl polychlorinates (PCBs) | Corticosterone levels, liver fibrosis | Mice, male [30] |
| Bisphenyl polybrominated (PBBs) | Fecundit cells, liver steatosis | Mice [31] | |
| Per- and polyfluoroalkyl substances (PFOS) | Liver injury, PFASs | mice [32] | |
| Dioxins | Cell-mediated immunity, liver injury | Human, mice [33] | |
| Plasticizers | Bisphenol A (BPA) | Fertility, liver steatosis | Mice, male female [34] |
| Phthalates | Insulin resistance and type II diabetes, overweight and obesity, liver steatosis | Male, female and children [35] | |
| Di(2-ethylhexyl) phthalate (DEHP) | Liver injury | Mice [36] | |
| Pesticides | Cypermethrin (CYP), atrazine (ATZ) | liver injury, growth parameters | Mice [37] |
| Dichlorophenyltrichloroethane (DDT) | neonatal body weight, liver damage | Mice, male, children [38] | |
| Permethrin | Dopamine transport, fatty liver | Mice, human [39] | |
| Drugs | Diethylstilbestrol (DES) | Expression of PDGF receptor, neonatal body weight, fatty liver | male and female/mice [40] |
| Heavy Metals | Arsenic | apoptotic index, liver injury | Mice [41,42] |
| Cadmium | Expression of metallothionein, pS2/TFF1, liver steatosis | Mice [43,44,45] | |
| Mercury | Growth, hepatic steatosis | Mice, human [44] |
EDCs have been identified as compounds that mimic or block the action of estrogen receptors, androgens, and thyroid hormones, and, among them, BPA is considered a weak xenoestrogen [26].
While detectable in amniotic fluid, placental tissue, umbilical cord blood, and breast milk, determining the minimum toxic dose of BPA required to exert harmful effects remains challenging [9].
Despite its low lipophilicity and rapid degradation (with a half-life of 4–5 h), recent studies suggest a potential bioaccumulation in adipose tissue, due to BPA’s high affinity for fatty acids [46]. Corroborating this possibility, a further study demonstrated that, after a BPA-free diet for one month, a group of patients with NAFLD exhibited a reduction in circulating plasma BPA levels without a significant reduction of urinary levels compared with baseline [10]. This event could be a result of BPA release from adipose tissue, which would explain the reduction of BPA in plasma but not in urine after the BPA-free diet. Nonylphenols (NPs), also known as alkylphenols, are found in various products, including lubricants in oil additives, laundry detergents, emulsifiers, and solubilizers [47]. Alongside these chemicals, pesticides fall within the POPs category [48]. These substances exhibit a relatively slow degradation rate and high lipophilicity, resulting in their widespread presence in organic tissues, particularly adipose tissue. Being semi-volatile, POPs can disperse across long distances via air and ocean currents [49].
2. The Effects of Obesogens across the Lifespan
In 2009, Green and Blumberg highlighted that certain EDCs function as “obesogenic” substances (i.e., non-steroidal estrogens, organotins, parabens, phthalates, polychlorinated biphenyls and bisphenols), actively promoting obesity by altering endocrine homeostasis. They proposed that such substances modify homeostatic mechanisms critical to weight control, thereby increasing individuals’ susceptibility to obesity, even when they maintain a healthy diet and exercise routine [49]. The obesogenic hypothesis for EDCs rests on two pivotal points: first, there is an increased susceptibility to obesity, which starts in the prenatal period and continues across the first years of life and, secondly, this susceptibility to obesity is boosted by a specific subclass of EDCs, which alters the programming of human development, influencing weight control later in life [50]. Consequently, “obesogens” are functionally defined as chemicals that promote obesity by increasing the number of fat cells, and/or by enhancing fat accumulation in existing adipocytes. Recently, the domain of obesogens has broadened, acknowledging that certain EDCs exert organ-specific effects, promoting the onset and progression of morbidities that are syndemic with obesity. However, these organ-specific effects are not mediated by an increase in adiposity [51]. For instance, some EDCs induce, in both human and animal models, hepatic steatosis associated with metabolic dysfunction (MASLD) not only through epigenetic programming during early prenatal and postnatal life, akin to “classic” obesogens, but also by interfering directly with the functioning of hepatocytes [52]. There has been a recent transition to a new nomenclature for what was previously termed non-alcoholic fatty liver disease associated with metabolic syndrome. This new framework, relevant for children, integrates multifactorial considerations. However, before applying the diagnostic criteria for MASLD, it is crucial to rule out other causes of fatty liver disease, to avoid overlooking the possibility of a dual pathology [53]. The newly accepted nomenclature is hepatic steatosis disease (NAFLD) associated with at least one of the following conditions: excess adiposity (overweight/obesity or abdominal obesity), prediabetes or type 2 diabetes mellitus (T2DM), metabolic abnormalities (dyslipidemia, insulin resistance, alteration of the intestinal hepatic axis), as well as genetic predisposition and epigenetic events [54]. This new definition definitively closes the pathophysiological link of fatty liver with metabolic dysfunction and insulin resistance, reinforcing its role as cardiometabolic risk factors associated with MASLD. Hepatic MASLD is characterized by a broad spectrum of abnormalities ranging from simple steatosis to steatohepatitis associated with metabolic dysfunction (MASH), with the latter condition implicating lobular inflammation, swelling, and often fibrosis. To date, a multitude of genetic, epigenetic, and environmental MASLD modifiers have been reported, especially in the perinatal period and in the first years of life. However, diagnosis is often delayed due to its non-specific clinical manifestation, so that its discovery often occurs during routine examinations [53,54].
3. EDC and MASLD
3.1. Prenatal Life
The Developmental Origins of Health and Disease (DOHaD) theory emphasizes the existence of critical developmental windows during fetal stages, wherein environmental pressure can induce subtle changes in gene expression, tissue organization, or other levels of biological organization. These changes can lead to lasting dysfunction and heightened susceptibility to chronic disease [55]. Unlike birth defects or neonatal ailments, these dysfunctions often surface later in life, encompassing conditions such as obesity, NAFLD, and subsequently, MASLD [56]. Several studies have demonstrated that low birth weight (LBW) infants, born to mothers who experienced either excessive or deficient nutrition, are more prone to develop an increased susceptibility to various diseases in adult life. These include heart disease, high blood pressure, obesity, T2D, osteoporosis, dyslipidemia, impaired glucose metabolism, and NAFLD [54,55,56,57].
A recent prospective cohort study examined 253 children within a pediatric population and found association between prenatal exposures to ubiquitous EDCs (notably organochlorine pesticides, PBDEs, PFAS, and metals) and increased liver damage and/or hepatocellular apoptosis. Among 1108 children from 6 different countries, 253 (22.8%) exhibited a high risk of liver damage, indicated by elevated transaminases. All children in the study showed exposure to various EDCs, as follows: 3 organochlorine pesticides, 5 polychlorinated biphenyls, 2 polybrominated diphenyl ethers (PBDEs), 3 phenols, 4 parabens, 10 phthalates, 4 organophosphate pesticides, 5 perfluoroalkyl substances, and 9 metals. The prevalence of liver injury varied among countries, with the highest prevalence in children from Greece (80 out of 253, 31.6%) and the lowest in children from Lithuania (11 out of 253, 4.3%). Additionally, there was an escalated likelihood of liver injury corresponding to increased quartiles of exposure to organochlorinated pesticides (odds ratio (OR): 44.95, 1% credibility interval (CrI): 21.1–71.1), PBDEs (OR: 57.95, 1% CrI: 34.1–84.1), perfluoroalkyl substances (OR: 73.95, 1% CrI: 45.2–09.2) and metals (OR: 21.95, 1% CrI: 65.3–02.0) [25].
Several studies have shown that a range of chronic conditions, including obesity, chronic diseases, but also lifestyle habits, diet and physical activity, T2D and NAFLD, can be correlated with epigenetic modifications occurring in cells and tissues during development. These alterations affect impaired tissue development, arising from early environmental factors such as stress, pharmaceuticals, nutrition, and environmental chemicals. Importantly, the effects of diseases induced during development might not become immediately apparent but rather manifest later in life [57] (Figure 2).
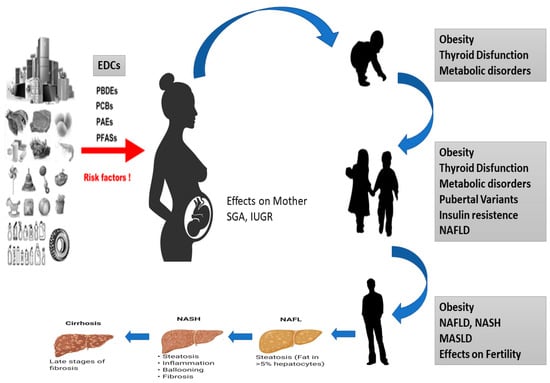
Figure 2. Effects of EDCs during development of MASLD in children. Diagram of how EDCs can affect the fetus, which can be born SGA or IUGR and is exposed to a series of epigenetic, genetic and environmental factors that can cause thyroid disease, obesity, metabolic disorders and NASH to develop in all stages of life.
Therefore, environmental monitoring data appear insufficient to predict population contamination levels and do not allow the detection of metabolite-related toxicities during fetal life. The experimental difficulties are also represented by the lack of knowledge of biological cause–effect relationships, which precludes the possibility of predicting the adverse effects of EDCs.
3.2. Young Adult Life
The liver stands as a crucial defense against harmful substances, employing complex and sophisticated mechanisms that include filtration, oxidation, and conjugation of chemical compounds. Enzyme systems such as cytochrome P450 (CYP) and UDP-glucuronosyltransferase (UGT) are responsible for eliminating more than 90% of these substances. While the complete metabolic pathways of toxins and the exact mechanisms by which EDCs disrupt liver function remain incompletely understood, it must be considered that their entry into the liver often coincides with food intake or contact with harmful materials. Consequently, the absorption and transport of EDCs often occurs alongside dietary lipids via the portal circulation. These compounds, characterized by high lipophilicity, swiftly diffuse across cell membranes, gaining expedited access to target sites. Upon reaching the liver, activation of nuclear receptors (NRs) by EDCs disrupts hormone signaling pathways and modulates the cytochrome P450 (CYP) system. This enzyme system comprises several isoforms, with CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A being the major players in toxin metabolism [52,58]. The influence of EDCs on this detoxification system remains a subject of debate. Studies have indicated that pesticides, parabens, phthalates, and BPA can reduce the catalytic efficiency of CYP450. These findings suggest that altered metabolism, coupled with prolonged exposure, significantly contributes to the bioaccumulation of these substances [59]. This intricate interaction can also convert some EDCs into more active metabolites. For instance, low molecular weight phthalates, such as di-(2-ethylhexyl) phthalate (DEHP), dimethyl phthalate (DMP), and dibutyl phthalate (DBP), can bio-transform into hydrolytic monoester metabolites with enhanced activity. An effective method to assess the impact of EDCs on liver function involves the use of traditional markers such as liver enzymes. Exposure to various POPs, including PCBs, occhlorinated dibenzodioxin (OCDD), and specific pesticides, has been linked to elevated bilirubin, ALT, and ALP levels. These elevations indicate a detrimental effect on liver function due to prolonged exposure to these pollutants in young adult populations [52,60,61,62].
It is therefore certain that individual genetic susceptibility, comorbidities and dietary habits can modify the impact of EDCs on the body. As examples on might look at the way a family history of T2DM or obesity may accentuate the diabetogenic effects of certain molecules; how a high-fat diet may increase the exposure to lipophilic EDCs; how EDCs modulating steroids’ action can give different results (depending on hormonal background, stage of body’s development, timing of exposure); and, finally, how trans-generational effects are possible. An important limitation to the interpretation of cross-sectional epidemiological studies is the lack of information on exposure to EDCs and possible confounding compounds. This does not allow one to establish causal links between exposure and effect.
4. Endocrine Disruptors and the Development of MASLD
Numerous EDCs (BPA, phthalates, PFOA and PFOS) are classified as “obesogenic”. The hypothesis put forth by Grun and Blumberg primarily focuses on the impacts of these exposures on adipocytes, leading to inflammation, oxidative stress, and effects on pancreatic β-cells. Consequently, this impact alters insulin secretion and contributes to the initiation and exacerbation of insulin resistance [63,64].
Endocrine disruptors possess the capacity to act as agonists or antagonists by binding to NRs. The liver harbors a vast array of NR [64]. The activation of classical NR activity typically involves ligand-mediated attachment to response elements situated in the promoter region of target genes. This is followed by binding of steroid receptor coactivator complexes (SRCs), recruiting additional regulators with diverse histone-modifying enzyme activities [65]. This extranuclear signaling leads to the activation of kinases and downstream signaling pathways that elicit biological responses irrespective of the nuclear positioning of NRs. Non-genomic signaling, characterized by its rapid action, occurs independently of RNA or protein synthesis [66].
Numerous NRs actively participate in non-genomic signaling. The most important of these are the steroid hormone receptors: estrogen receptors (ERα and ERβ), androgen receptors (ARs), and progesterone receptors (PRs). Additionally, other NRs, such as peroxisome proliferator-activated γ receptor (PPARγ), retinoid α X receptor (RXRα), thyroid α receptor isoforms (TRα), and retinoic receptor isoforms (RARα and RARγ), seem to employ similar signaling mechanisms. Retinoid X receptors (RXRα, RXRβ, and RXRγ), which include α, β, and γ receptors, are activated by peroxisome proliferators (PPARs), liver X receptors α and β (LXR), farnesoid X α receptor (FXRα), and thyroid receptors (TR), and have been implicated in the modulation of MASLD [25,26,67]. For instance, LXRs, when bound to oxysterols, activate lipogenic genetic programs such as FAS and SREBP1, leading to lipid accumulation in the liver. FXR responds to bile acids by inducing the expression of bile acid export genes and NR0B2, which in turn induces the expression of SREBP1. FXR can reduce fatty liver disease and insulin resistance. FXR agonists such as obeticholic acid are currently undergoing clinical trials for the treatment of NASH in humans [68].
Studies have shown that FXR inhibits the expression of fatty acid synthetase (FAS) and reduces the synthesis of fatty acids and triglycerides. The mechanism for this is the suppression of sterol regulatory element binding protein-1c (SREBP-1c) by FXR via SHP-mediated inhibition of co-activator recruitment to SREBP1c promoters [68]. Several studies in mice have shown that, when combined with other MASLD risk factors, EDCs typically induce the NAFLD phenotype in exposed rodents. A recent comprehensive review of 371 studies suggests that 123 unique environmental chemicals are associated with NAFLD in rodents, with pesticides accounting for the majority (44%), while PCBs and dioxins were the most potent, based on levels [69]. Two baseline studies have reported that perinatal exposure to BPA (50–100 μg/kg per day), combined with a high-fat diet (HFD) after weaning, induced more severe fatty liver disease and increased inflammation and mild fibrosis but that these effects were observed only in male subjects [69,70]. In vitro studies on HepG2 cells have highlighted BPA’s role in liver inflammation by triggering the release of proinflammatory cytokines (IL-8 and TNF-α). Acaroz et al. have reported increased levels of TNF-α, IL-6, and IL-1β, alongside decreased levels of IL-10, after low oral BPA doses, promoting the development of pro-inflammatory micro-environments and dose-dependent histopathological changes in rat livers [70].
These findings suggest that, apart from increasing hepatic lipid accumulation, exposure to EDCs may trigger inflammatory infiltration, exacerbating the development of NASH and MASLD [56,70]. The observed increase in hepatic lipid accumulation linked to BPA utilization might result from an imbalance in free fatty acid (FFA) absorption, synthesis, or β-oxidation and/or the export of triglycerides as very low density lipoprotein VLDL [56].
The impact of EDCs on long-term health, notably their capacity to alter the epigenome, has been extensively studied. These disruptions can induce lasting alterations in the epigenome which, due to the hereditary nature of epigenetic programs, can persist across many cell generations and throughout an individual’s life [71]. DNA methylation, the first molecular mechanism identified for the epigenetic regulation of gene expression, occurs through the enzymatic transfer of a methyl group to cytosine bases of DNA, resulting in the formation of 5-methylcytosine [70]. In animal models, several studies have explored how EDCs might interfere with DNA methylation in liver diseases [70]. In terms of pathologic findings, mice and rats treated with BPA showed dose-dependent dilation of liver tissue sinusoids, congestion, inflammation, and necrosis [71]. Prenatal BPA exposure has been associated with the metabolic health of offspring. In fact, such exposure has been shown to alter gene expression profiles and cause peripheral insulin resistance and hepatic lipotoxicity [72]. Rodent models further support the idea that gestational exposure to BPA can promote the development of NAFLD by perturbing the activity of nuclear transcription factors.
This entry is adapted from the peer-reviewed paper 10.3390/metabo14010071
This entry is offline, you can click here to edit this entry!