1. Introduction
Alcohol use disorder (AUD) and major depressive disorder (MDD) are the most common co-occurring disorders substantially burdening worldwide well-being socially and financially [
1,
2,
3]. Notably, AUD and MDD share many neurobiological underpinnings of symptoms [
4,
5,
6,
7,
8,
9,
10,
11]. Among several notable biological factors attributed to the pathophysiology of AUD and MDD, the dysregulation of γ-aminobutyric acid (GABA) levels and GABA receptor-mediated signaling are crucial to understanding specific phenotypes and, thereby, therapeutic targets [
12,
13,
14]. As the primary inhibitory neurotransmitter in the brain, GABA is involved in multiple crucial brain functions such as learning and memory, motor coordination, emotion regulation, sensory processing, addictive behavior, and circadian rhythm [
15,
16,
17,
18,
19,
20,
21]. In MDD patients, the biomarkers associated with GABA metabolism and signaling are altered in the cortical region [
22]. While it is known that brain GABA deficits are attributed to the etiology of MDD, GABAergic involvement in the molecular mechanisms of AUD is less clear. Chronic alcohol administration decreases GABA-mediated responses in the cortex and nucleus accumbens, while alcohol increases the GABA released into the central nucleus of the amygdala, facilitating GABAergic transmission [
23]. Furthermore, alteration in GABAergic signaling has been implicated in alcohol reinforcement effects and compulsive drinking in variable ways [
24,
25]. While the activation of GABA
B receptors is known to decrease the reinforcement actions of alcohol, acute blockade of GABA
A receptors can block the motivation for responding to alcohol [
26,
27,
28,
29]. GABA and GABA receptors have also been involved in the changes in the reward system associated with acute withdrawal [
13,
14].
2. Astrocytic Regulation of GABA in the CNS
GABA exerts its inhibitory effects by interacting with two distinct receptor types: GABA
A receptors (GABA
AR) and GABA
B receptors (GABA
BR). GABA
AR is an ionotropic receptor and ligand-gated ion channel responsible for inhibitory synaptic transmission in the CNS [
45,
46,
47]. GABA
BR, a metabotropic receptor, operates at a slower pace through G-protein-coupled mediated signaling [
48]. Immunohistochemical analysis of the adult human brain reveals that astrocytes express GABA
AR and GABA
BR at levels comparable to or even more significant than those observed in known GABAergic neurons. Additionally, cultured astrocytes derived from adult human brain tissue confirm the presence of GABA
AR and GABA
BR at both the mRNA and protein levels, establishing their dual GABAergic and GABAceptive characteristics [
49]. In primary cell cultures and rodent slices, extracellular GABA can activate astrocytic GABAARs, increasing Cl
− concentrations in astrocytes. GABA also activates astrocytic GABA
BRs, mediating slow inhibitory signaling in the brain via the activation of Gi/o-type G-proteins, leading to the inhibition of adenylyl cyclase. The activation of astrocytic GABA
BRs increases intracellular Ca
2+, which triggers the release of Ca
2+ from the intracellular pools. Ca
2+ oscillations in astrocytes alter glutamate release and GABA transporter expression. Therefore, astrocytes can internalize GABA via GABA receptors and transporters, indicating their GABAceptive properties [
50].
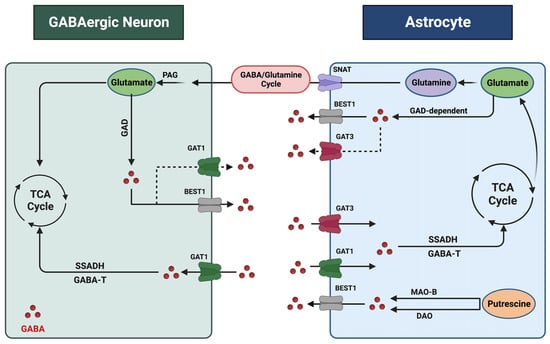
The regulation of GABA levels within the CNS is contingent upon the dynamic interaction between neurons and astrocytes (
Figure 1). Both neurons and astrocytes are responsible for the synthesis, release, and reuptake of GABA within synapses. Each process responds to various pathophysiological stimuli to maintain GABA homeostasis. These processes are multifaceted, with distinct mechanisms operating in different cell types and regions of the brain [
30,
51,
52,
53,
54,
55]. Due to their close association with neurons and ability to engage in GABA synthesis, release, and reuptake, astrocytes are now considered pivotal contributors to the intricate task of regulating GABA homeostasis within the CNS [
56]. Two glutamate decarboxylases, 1 and 2 (GAD1 and GAD2), produce GABA from glutamate. GAD1 is primarily expressed in neuronal cell bodies, while GAD2 is found in axon terminals [
57]. Notably, GAD1, which plays a vital role in GABA synthesis, has also been observed in astrocytes [
50,
58]. Another crucial precursor of GABA in the CNS is putrescine, metabolized by monoamine oxidase B (MAO-B) and diamine oxidase (DAO). MAO-B is particularly prevalent in astrocytes, especially in cerebellar and striatal astrocytes [
59]. Recent studies strongly suggest that GABA synthesis in astrocytes is a predominant factor in regulating GABA levels, suggesting that astrocytes are GABAergic cells [
50].
Figure 1. Astrocyte–GABAergic neuron interaction. GABA undergoes extensive recycling between neurons and astrocytes, a process coordinated and regulated by cell-specific transporters and enzymes. Within astrocytes, GABA and glutamate are metabolized, supporting the synthesis of the non-neuroactive amino acid glutamine. Astrocyte-derived glutamine is taken up by neurons and converted to glutamate by phosphate-activated glutaminase (PAG) to replenish the neurotransmitter pool. Sodium-coupled neutral amino acid transporters (SNATs) facilitate glutamate transfer. Astrocytes synthesize GABA from glutamate through a glutamate decarboxylase (GAD)-dependent pathway. Putrescine serves as another precursor for GABA, and monoamine oxidase B (MAO-B) and diamine oxidase (DAO) are the key enzymes in this pathway. Additionally, the BEST1 (bestrophin 1) channel mediates GABA release. GABA transporters recycle excess GABA from the synapse through uptake into neurons and astrocytes. Astrocytes express GAT1 and GAT3, while GABAergic neurons express GAT1. Following uptake, intracellular GABA is metabolized by the enzymes GABA transaminase (GABA-T) and through the intermediate succinate semi-aldehyde by the enzyme succinate semi-aldehyde dehydrogenase (SSADH). This figure was created with
BioRender.com (accessed on 17 November 2023).
The release of GABA from astrocytes occurs through three distinct mechanisms: calcium-dependent vesicular exocytosis, direct release into the extracellular space via GABA transporters (GATs) in reverse mode, or through GABA-permeable channels [
51,
60,
61]. GATs function as secondary active electrogenic transporters and utilize sodium and chloride ion exchange with GABA uptake. Although the primary role of GATs is to remove excess GABA from the extracellular space, they were found to act in reverse mode, releasing GABA into the extracellular space in certain conditions, a function that remains controversial [
56,
62]. GATs are found in presynaptic neurons and astrocytes, with four known types (GAT1, GAT2, GAT3, and BGT1), among which GAT1 and GAT3 exhibit high affinity for GABA [
61,
62]. While GAT1 was thought to be primarily expressed in neurons, recent research shows that GAT1 is expressed in cortical and thalamic astrocytes as well. [
63,
64]. GAT3, on the other hand, is exclusively expressed in astrocytes predominantly localized at the astrocytic processes, modulating tonic inhibitory currents in postsynaptic cells. GAT3 activities influence various astrocytic functions encompassing the regulation of inhibitory synapse efficacy, excitatory neurotransmission, and astrocyte synaptic proximity, underscoring the role of GAT3 as a key glial GABA transporter [
65,
66]. Interestingly, the genetic ablation of GAT3 is lethal in mice, suggesting its critical role in GABA homeostasis during early embryogenesis and development [
67].
Furthermore, astrocytic GABA release is facilitated by permeable membrane channels, such as BEST1, which is a calcium-dependent anion channel and is considered a vital mechanism of astrocytic GABA release [
51]. Astrocytic GABA is known to interact with mainly surrounding neurons [
50]. A recent study shows that astrocytes sense environmental change and release gliotransmitters, including GABA, to alter neuronal activities [
68,
69]. Astrocytic GABA can interact with synaptic and extrasynaptic receptors [
60,
68].
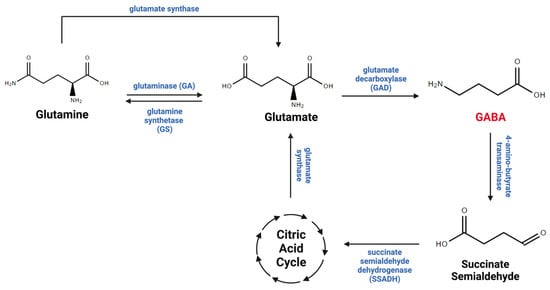
The GABA taken up by astrocytes via GABA transporters is either recycled into the GABA/glutamine cycle, which plays critical roles in GABA metabolism between neurons and astrocytes (
Figure 2), or metabolized within mitochondria by the GABA transaminase (GABA-T) enzyme, which is present in both neurons and astrocytes [
53,
70]. Importantly, research has demonstrated that almost half of the released GABA is taken up and metabolized by astrocytes, underscoring their role in regulating GABA levels in the CNS [
67]. Therefore, it is evident that astrocytes are considered essential cells in maintaining GABA homeostasis in the brain.
Figure 2. GABA metabolism. GABA is metabolized through the GABA/glutamate/glutamine cycle, which connects neurotransmitter homeostasis and cellular energy metabolism. Glutamate is the principal biological precursor for GABA through the enzyme glutamate decarboxylase (GAD). GABA is converted to succinic semi-aldehyde, which is metabolized into the tricarboxylic acid (TCA) cycle through the enzyme succinic semi-aldehyde dehydrogenase (SSDAH). GABA, glutamate, and glutamine undergo oxidation within the TCA cycle, actively contributing to energy production in neurons and astrocytes. This figure was created with
BioRender.com (accessed on 17 November 2023).
3. Astrocytes and GABA in Alcohol Use Disorder (AUD)
Alcohol use disorder (AUD) is a chronic, relapsing disease characterized by compulsive drug seeking despite negative consequences on an individual’s life [
71]. Alcohol (or ethanol) exerts its toxicity through alterations in multiple neurotransmitter systems, including the GABA, serotonin, dopamine, glutamate, acetylcholine, and opioid systems [
72]. The neurotransmitter imbalances result in malfunctioning brain circuits responsible for cognitive function, decision making, motivation, reward, affect, and the stress response [
73]. Despite the dire health and psychosocial consequences, AUD continues to persist as one of the leading causes of death globally [
74,
75]. According to the most recent report from the National Institute of Health (NIH), the annual mortality rate due to alcohol-related causes exceeds 140,000 individuals, making it one of the leading four preventable factors contributing to fatalities in the United States [
76]. AUD is typically associated with the development of tolerance, dependence, and the impairment of social and occupational functioning [
77]. Recent research has suggested a significant role in the facilitation of GABAergic transmission in the addictive properties of alcohol [
13,
14,
78]. It has been proposed that GABA plays a substantial role in the neuroadaptations linked to the progression from controlled alcohol consumption to excessive drinking characterized by dependence and relapse [
79,
80].
Evidence indicates that alcohol alters GABA-mediated responses in various brain regions, including the cortex and substantia nigra [
81]. Recent studies have demonstrated that alterations in GABA transmission are particularly pronounced in regions implicated in the negative reinforcing aspects of alcohol, such as the ventral tegmental area (VTA), globus pallidus (GP), and the amygdala [
82,
83]. In experiments involving in vitro slice preparations from the central amygdala, acute alcohol exposure enhances GABA
A inhibitory postsynaptic currents. In rats chronically exposed to alcohol, an increase in evoked GABA release was observed [
84]. Additionally, acute pharmacological inhibition of GABA
AR function effectively diminishes the motivation for responding to alcohol [
13].
Furthermore, the utilization of selective GABAB agonists has been shown to reduce alcohol self-administration in rats and mitigate the alcohol deprivation effect in alcohol-preferring rats [
85]. Additional experiments have revealed that the combination of agonists and antagonists at the GABA
AR benzodiazepine site leads to a noteworthy reduction in alcohol administration when administered into the amygdala [
86,
87,
88].
Moreover, research employing pharmacological GABA agonists and antagonists has implicated the GABA system in both the physical and affective symptoms associated with alcohol withdrawal. GABA agonists were found to have the capacity to reduce CNS hyper-flexibility during alcohol withdrawal-induced seizures [
89,
90,
91]. Consistently, GABA mimetics potentiate the sedative and motor effects of alcohol, an effect that was counteracted by GABA antagonists [
24]. However, the underlying molecular mechanisms of the central effects of alcohol involving GABA-mediated signaling remain unclear. Notably, recent evidence has highlighted the role of astrocytes in modulating GABA transmission within the brain [
30]. Chronic alcohol exposure alters the balance between inhibitory and excitatory neurotransmissions in various brain regions such as the cortex and the striatum [
92]. Remarkably, a single astrocyte can modulate up to one million inhibitory and excitatory synapses. Consequently, any disruption to even a small subset of astrocytes can profoundly impact on the delicate balance between excitation and inhibition, ultimately, affecting brain function and behavior [
93]. However, additional research may elucidate the precise role of astrocytes in ethanol-induced GABAergic neurotransmission.
An essential aspect of AUD is the disruption of the balance between goal-directed and habitual reward-seeking behaviors. Alcohol is known to exert effects on several signaling systems in the cortico-striatal circuits that may collectively contribute to the impairment of behavioral flexibility and motivate the transition from goal-directed to habitual alcohol drinking [
94]. Changes in GABA release, uptake, and GABA receptor signaling across chronic alcohol exposure are critically involved in the acquisition of both goal-directed and habitual behaviors [
95]. Studies indicate that chronic alcohol exposure may reduce the expression of GABA
AR in the dorsal striatum, a region crucial for forming and expressing stimulus–response habits [
96,
97]. However, the precise direction in which alterations in GABA release and signaling drive behavior is unknown.
Given the pivotal role of astrocytes in regulating GABAergic transmission, recent studies have investigated the specific role of astrocytic GABAergic signaling in cognition and behavior. Recently, researchers documented that genetic ablation of GABA
BR in the medial prefrontal cortex astrocytes altered the low gamma oscillations and firing properties of cortical neurons, affecting goal-directed behaviors [
98].
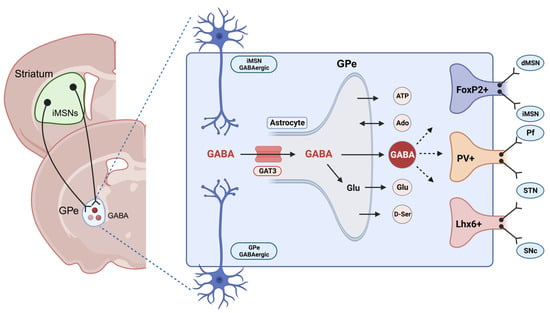
Within the striatopallidal circuits, the dorsomedial striatum (DMS) and dorsolateral striatum (DLS) are the primary neural regions responsible for regulating goal-directed and habitual behaviors, respectively [
99,
100,
101]. Nevertheless, the external globus pallidus (GPe), an area known to contain a substantial population of astrocytes, assumes a pivotal role in facilitating and coordinating the neurotransmission between the DMS and DLS, making it an integrative center for modulating the flexibility of reward-related behaviors (
Figure 3) [
102,
103]. Notably, extrasynaptic neurotransmitters can trigger astrocyte Ca
2+ signaling, and reciprocally, astrocyte Ca
2+ signals modulate the function of the neural circuits through various gliotransmitters. Our recent research demonstrated that chemogenetic activation of astrocytes in the DMS differentially regulated striatal medium spiny neuron (MSN) activities and induced a shift from habitual to goal-directed reward-seeking behavior [
101,
104]. Despite these findings, it has not yet been fully elucidated how astrocytic modulation in the basal ganglia circuit may govern neuronal activities associated with goal-directed and habitual reward-seeking behavior in AUD. While the general role of astrocytes in regulating behavior, excitatory/inhibitory balance, and neuroplasticity in different brain regions is widely recognized, the specific involvement of astrocytic GABA signaling in the context of AUD remains unclear.
Figure 3. GABA transport in the globus pallidus externus (GPe) neurons. The “GPe” accommodates diverse neuronal populations, such as parvalbumin+ neurons and arkypallidal neurons expressing FoxP2+ and LIM homeobox-positive (Lhx6+) neurons. GABAergic neurons in the GPe receive inputs from the striatal indirect pathway medium spiny neurons (iMSNs). The GPe comprises numerous astrocytes capable of releasing neurotransmitters such as ATP, adenosine, glutamate, D-serine, and GABA [
82,
83]. GABA transporter 3 (GAT3) is an astrocyte-specific transporter responsible for either releasing into the synapse or metabolizing into the GABA/glutamine cycle. This figure was created with
BioRender.com (accessed on 17 November 2023).
Our recent study has revealed that GPe astrocyte activity is suppressed during habitual learning in mice. Notably, chemogenetic activation of astrocytes has been shown to reduce habitual behaviors while concurrently enhancing goal-directed reward-seeking behaviors in operant conditioning experiments. Additionally, we found that activation of astrocytes reduced the overall activity of GPe neurons, which facilitated the transition from habitual to goal-directed alcohol-seeking behavior as well. Intriguingly, we observed an increase in GAT3 mRNA levels during habit formation, and the selective inhibition of GAT3 reversed the impact of astrocytes on the transition from habitual to goal-directed alcohol seeking. Our finding indicates that the upregulation of GAT3 in the GPe may deactivate astrocytes, diminishing their inhibitory influence on GPe neurons. These findings underscore the potential essential role of astrocytic GAT3 in regulating reward-related behavioral flexibility within the GPe [
105]. Recent evidence supports the idea that GAT3 can govern astrocytic activity [
106]. Nevertheless, the underlying mechanisms of GAT3 signaling, potentially involving other neurotransmitters, remain an area yet to be thoroughly explored. Moreover, given the diverse neuronal populations within the GPe, such as parvalbumin-expressing neurons, arkypallidal feedback, and prototypic feedforward neurons (
Figure 3), further investigations on the specific changes related to GAT3 activity within distinct GPe cell types are warranted.
GABA transporters have recently garnered increasing attention within the field of addiction research, with a particular focus on GAT3, known for its high expression in astrocytes [
66,
107]. A recent study has uncovered a decrease in GAT3 mRNA levels in the amygdala, a forebrain structure recognized as a central hub for GABAergic influences on alcohol reinforcement, in alcohol-preferring mice. A reduction in several GABAAR subunits accompanied this decrease. The researchers proposed that this latter observation could indicate heightened GABAergic activity resulting from a decrease in the extracellular clearance of GABA. To further substantiate these findings, the researchers employed a viral GAT3 (
Slc6a11) knockdown strategy in a mice group that initially preferred saccharin over alcohol. Their results revealed that, following the full expression of the injected virus in the amygdala, mice with GAT3 knockdown exhibited a shift in behavior from saccharin preference to alcohol preference [
78].
The prolonged administration of alcohol, sufficient to induce dependence and escalate alcohol consumption, which is linked to increased GABA release within the amygdala, is accompanied by enhanced sensitivity to GABA agonists [
108,
109]. A study demonstrated that the microinjection of the GABAA agonist muscimol into the central nucleus of the amygdala (CeA) of alcohol-dependent rats reduced alcohol self-administration. However, this effect was not observed in alcohol nondependent rats [
23,
110]. Furthermore, a recent study revealed that rats with diminished GAT3 expression in the amygdala exhibited a propensity for persistent alcohol seeking, even when the alcohol was mixed with quinine. These findings suggest that extracellular GABA homeostasis in the amygdala plays a vital role in vulnerability to compulsive alcohol seeking. On the other hand, baclofen, a GABA
BR agonist, lowers extracellular GABA levels in the amygdala and reduces alcohol consumption in mice and humans [
111]. Interestingly, baclofen’s efficacy in diminishing the susceptibility to compulsive drinking and GAT3 expression in the amygdala are inversely correlated. This study showed a positive correlation between GAT3 mRNA levels in the amygdala and increased resistance to quinine in baclofen-treated but not vehicle-treated rats. This implies that baclofen’s effects may be mediated by normalizing impaired GABA clearance resulting from low GAT3 expression in the amygdala. However, it remains unknown whether solely restoring GABA homeostasis in the amygdala is sufficient to reverse compulsive alcohol seeking. Furthermore, additional research may be necessary to elucidate the psychological consequences of manipulating the GABAergic system within the amygdala and whether GAT3 expression is associated with baclofen’s known side effects. Collectively, these findings highlight the pivotal role played by astrocytic GAT3 in the molecular mechanisms of alcohol-seeking behavior and its potential as a therapeutic target for AUD.
This entry is adapted from the peer-reviewed paper 10.3390/cells13040318