Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medicine, Research & Experimental
Phosphatase and tensin homolog (PTEN) is a tumor suppressor due to its ability to regulate cell survival, growth, and proliferation by downregulating the PI3K/AKT signaling pathway. In addition, PTEN plays an essential role in other physiological events associated with cell growth demands, such as ischemia-reperfusion, nerve injury, and immune responsiveness.
- PTEN
- redox regulation
- oxidative inhibition
- ROS
- cell signaling
1. Cardiovascular Remodeling
Studies indicate the involvement of the serine/threonine kinase AKT as a mediator in the process of ischemic preconditioning, a short transient period of sustenance during ischemia-reperfusion injury [51,52,53,54]. In ischemic preconditioning, AKT signaling is upregulated and prevents cardiomyocytes from undergoing apoptosis [53,54,55,56]. The PI3K/AKT/mTOR pathway plays a significant role in protecting against ischemia-reperfusion injury, particularly in the context of ischemic preconditioning in cardiac tissue. Accordingly, reversible PTEN downregulation has been suggested as a viable therapeutic approach to mitigate ischemia-reperfusion-related cardiac damage [57]. A study revealed that PTEN plays a pivotal role in the post-myocardial infarction remodeling process: Partial PTEN inactivation, by regulating the AKT signaling pathway, can increase interleukin IL-10 and consequently decrease tumor necrosis factor TNFα and matrix metalloproteinase MMP2 expression in the heart. However, the authors were not able to determine the exact source of generated IL-10, apart from immune cells. It probably comes from endothelial cells and fibroblasts [58]. Several research studies demonstrate that IL-10 can eventually attenuate apoptosis and facilitate cardiac remodeling after myocardial infarction [59,60,61,62]. Hence, PTEN inhibition could be an effective approach for improving cardiac conservation after ischemia [63,64].
During acute myocardial infarction, the heart suffers from oxidative stress with increased ROS levels [23]. In the acute and chronic cellular response to this event, NOX2 is overexpressed in human cardiomyocytes, which may not interfere with the activity of macrophages [65,66,67]. Since PTEN oxidation is likely to occur near the site of ROS formation and both PIP3 and the NOX complex are located in the plasma membrane, H2O2 generated from NOXs is the primary candidate for inhibiting the PI3K/AKT pathway via PTEN oxidation. There is substantial supporting evidence indicating that elevated PIP3 signaling contributes to the activation of the NOX complex in both phagocytic and non-phagocytic cells. The increase in PIP3 levels is proposed to be a key factor in initiating the activation of the NOX complex [41,43]. This may create a circular impact, where ROS generated from NOXs can inhibit PTEN and enhance the PI3K/AKT pathway, which, in turn, promotes NOX activity.
Cai and Semenza were the first to describe the modulation of PTEN during ischemia-reperfusion injury. During the first 15 min of ischemia, PTEN undergoes dephosphorylation and proteasomal degradation. However, the kinetics reveal that not all PTEN activity is impaired during this initial phase and AKT phosphorylation increases without any significant changes. This indicates that the dephosphorylation and degradation of PTEN do not greatly hinder its function. However, in the subsequent initial phase of reperfusion, there is a notable increase in oxidized PTEN and, consequently, phosphorylated AKT. Their findings clarify that the surge in AKT phosphorylation during this short reperfusion period is caused by the oxidative inhibition of the remaining PTEN [68]. Simultaneously, elevated levels of ROS have been observed in both injured cardiomyocytes and intact hearts during ischemia-reperfusion events [68,69]. Therefore, the oxidation of PTEN during the initial reperfusion period is related to the concurrent rise in ROS levels. (Figure 1).
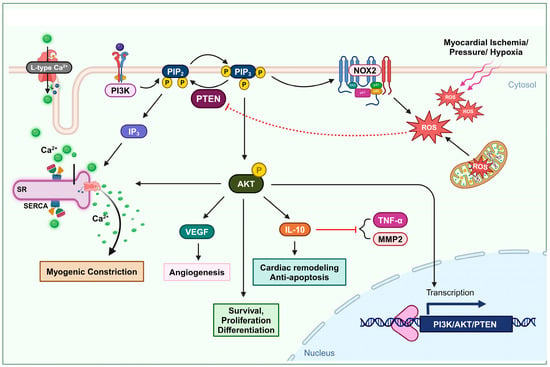
Figure 1. Oxidation of PTEN in cardiovascular remodeling and myogenic constriction. Ischemia or elevated blood pressure conditions induce the production of ROS. These ROS deactivate PTEN, leading to an increase in the AKT signaling pathway. The activation of the AKT pathway enhances cell survival, proliferation, and differentiation. Furthermore, PTEN-mediated AKT activation upregulates IL-10 expression, promoting cardiac remodeling and preventing apoptosis. It also elevates VEGF expression, facilitating angiogenesis. This mechanism also involves L-type calcium channel activity and the formation of IP3, which stimulates Ca2+ secretion, thus increasing intracellular Ca2+ levels and promoting myogenic constriction.
One vital mechanism of injured tissue in cases of blood supply shortage, due to ischemia or infarction events, is angiogenesis. Angiogenesis is defined as the formation of new blood vessels [70]. Vascular endothelial growth factor (VEGF) is associated with promoting angiogenesis. Upregulation of VEGF can be a potential treatment approach to induce axonal outgrowth and following angiogenesis after cerebral ischemia [71], as well as to restore blood flow in ischemic tissues after myocardial infarction [72]. Experimental data reported by Connor et al. indicate that the overexpression of manganese superoxide dismutase (SOD2) increases the production of mitochondrial H2O2, which triggers angiogenic activity. In this process, mitochondrial H2O2 can oxidize PTEN and upregulate the PI3K/AKT signaling axis, subsequently activating VEGF production [73] (Figure 1).
2. Vascular Constriction
Accumulating evidence highlights the significant role of PI3K/AKT-dependent signaling pathways in various fundamental cellular functions within the cardiovascular system. These functions include processes such as the maturation and growth, mechanotransduction, contractility, and proliferation and migration of both cardiac and vascular smooth muscle cells [74,75,76,77,78]. Dysfunction of this signaling pathway plays an essential role in cardiovascular pathophysiological conditions, such as heart failure, atherosclerosis, and hypertension [79,80,81,82]. Wu et al. observed that in the rostral ventrolateral medulla of spontaneously hypertensive rats, ROS originating from NOXs and mitochondrial oxidative stress reduced the catalytic ability of PTEN via oxidation. Consequently, the ensuing activation of the PI3K/AKT signaling pathway may lead to neurogenic hypertension [82].
Maintaining a consistent cerebral blood flow distribution through myogenic tone development is vital for neurons, which lack glucose storage and rely solely on a continuous blood supply of glucose and oxygen for normal metabolic function and under conditions of increased demands [83]. The role of PI3K in mediating the impact of physical forces, such as pressure, shearing, and stretching, on vascular smooth muscle cells and various other cell types, is well recognized [84]. Gebremedhin et al. found that elevated intraluminal pressure in cerebral arteries leads to an increase in ROS generation, leading to the oxidative inactivation of PTEN. This, in turn, results in the upregulation of PI3K/AKT activity and the release of IP3. The activation of AKT can induce the inhibition of arterial calcium-activated potassium channels, membrane depolarization, and L-type calcium channels. In addition, the formation of inositol (3,4,5)-triphosphate (IP3) stimulates the sarcoplasmic reticulum to release Ca2+, resulting in an increase in intracellular Ca2+ levels and the initiation of pressure-dependent myogenic constriction in cerebral arteries [83] (Figure 1).
3. Neuro-Regeneration and Neuro-Survival
PTEN activity has been shown to substantially limit cell survival in the challenging context of cerebral ischemia [64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85]. Numerous studies have demonstrated that inhibiting PTEN to activate the PI3K/AKT pathway provides protection to the brain during stroke [86,87,88,89,90,91]. The reduction in the PI3K/AKT/GSK-3β/mTOR signaling pathway by neuronal PTEN impairs axon growth and nerve regeneration in both the peripheral and central nervous systems, post-neuronal injuries, and ischemic conditions. Strong evidence consistently supports PTEN’s inhibitory role in critical neurological processes in pathological contexts [92,93,94,95,96,97,98]. Enhancing the activity of the PI3K/AKT pathway has been shown to increase axon growth [99]. Therefore, it is clear that PTEN, an intrinsic inhibitor of the PI3K pathway, plays a significant role in regulating the growth of central axons. PTEN’s activity also impedes nerve regeneration following neuronal injury, which is crucial for neural function recovery [96]. Hence, deliberately inhibiting PTEN activity emerges as a strategically advantageous approach with pronounced benefits for facilitating neuronal regeneration following injury. Empirical evidence shows that deleting PTEN in the spinal cord or optic nerve significantly enhances nerve regeneration after injury [100]. Targeted application of local pharmacological agents to suppress PTEN or the precise utilization of siRNA-based techniques to specifically downregulate PTEN expression at injury sites serves as a potent and effective strategy for accelerating the intricate axon outgrowth process and expediting the overall neuronal recovery [101]. Even in genetic diseases, such as spinal muscular atrophy, managing protein synthesis in motor neurons via PTEN depletion could be a therapeutic strategy [102,103]. Experimental data demonstrate that ROS signaling plays an essential role in promoting the self-renewal, proliferation, and differentiation of neural stem cells and neural progenitor cells via a regulatory mechanism in which the oxidation of PTEN by ROS upregulates the PI3K/AKT signaling pathway [104].
After neuronal injury, the injured axons are exposed to a highly oxidative and inflammation-driven environment. Under these conditions, growth cones, which are crucial for axon extension, initially collapse and retract. This process involves the oxidation of actin and produces ROS [105]. In a study, two experimental models were used to investigate the role of ROS generation in neuronal death and the involvement of PTEN in neurodegenerative diseases. Oxygen–glucose deprivation and the neurotoxin 1-methyl-4-phenylpyridinium iodide were applied to neural cells to simulate cerebral ischemia and Parkinson’s disease. However, it was found that ROS generated under these conditions did not cause oxidative inactivation to all cellular PTEN, allowing PTEN to maintain its functional activity. The suggested explanation is that the deactivation of PTEN phosphatase by ROS requires suitable intracellular co-localization with the site where these ROS are actively produced [106].
Experimental data demonstrate that the presence of peroxynitrite can prevent etoposide-induced apoptotic cell death in primary cortical neurons. This effect is primarily due to the oxidation of PTEN and the subsequent upregulation of the PI3K/AKT signaling pathway. Although the anti-apoptotic implication of peroxynitrite is subject to dispute, these data concurrently strengthen the potential of PTEN oxidation in promoting neuroprotection [115] (Figure 2).
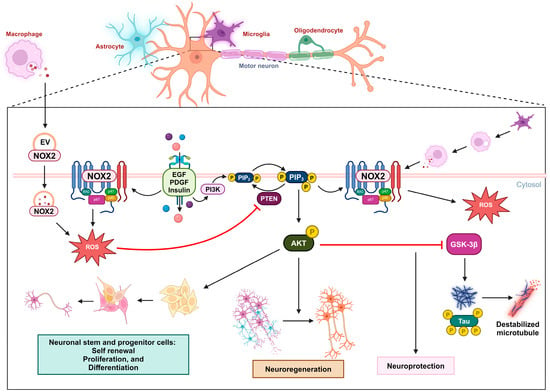
Figure 2. Oxidative inactivation of PTEN in nerve survival and regeneration. During neuronal injury, the NOX2-derived ROS concentration increases due to receptor kinase stimulation or extracellular vesicles released by macrophages. These ROS oxidize PTEN, leading to the activation of the PIP3/AKT signaling pathway, which promotes nerve regeneration. This mechanism can also promote self-renewal, proliferation, and differentiation in neuronal stem and progenitor cells. In the context of Alzheimer’s disease, the activation of the AKT pathway can downregulate GSK3β activity and the subsequent phosphorylation of the tau protein, providing neuroprotection.
3. Immune Responsiveness
Granulopoiesis is an emergency response to acute infection or inflammation, in which neutrophils are rapidly and massively produced and deployed from the bone marrow. Cytokines such as IL-6 and granulocyte colony-stimulating factor (G-CSF) are usually elevated during acute inflammation and may play a role in emergency granulopoiesis by inducing granulocyte differentiation [116,117]. In acute myocardial infarction, the myocardium also releases IL-6 and TNFα, and plasma levels of these cytokines increase after a brief episode of coronary artery blockage [118,119,120]. Kwak et al. demonstrated that an increase in ROS levels in the bone marrow alone is sufficient to trigger granulopoiesis. The elevated ROS concentration is important in promoting the proliferation and differentiation of myeloid progenitor cells via upregulated AKT signal transduction, which occurs due to the oxidative inhibition of PTEN’s phosphatase activity. During emergency granulopoiesis, these ROS are mainly produced by myeloid cells via phagocytic NOX2 activity, which can be induced by the cytokines G-CSF and TNFα. Therefore, the oxidative inactivation of PTEN by NADPH-oxidase-dependent ROS is an essential mechanism for prompting emergency granulopoiesis [121]. PI3K/AKT activity has also been shown to be a robust pivotal factor in the development of ROS-producing macrophages [122] (Figure 3).
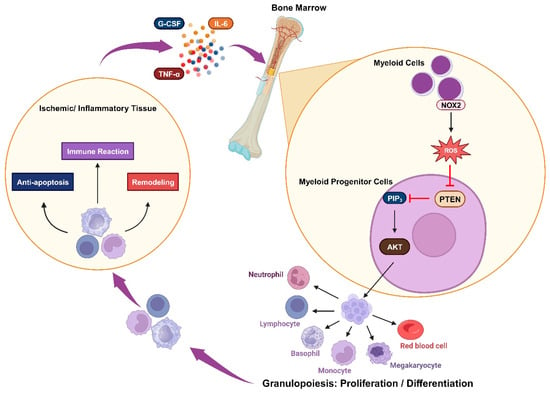
Figure 3. Oxidative inactivation of PTEN in immune responsiveness: Ischemia or inflammation can lead to elevated plasma cytokines, which stimulate myeloid cells to produce NOX2-derived ROS. These ROS mediate the AKT signaling pathway by inhibiting PTEN and trigger granulopoiesis, promoting the proliferation and differentiation of immune cells. These cells engage in immune reactions while also contributing to anti-apoptosis and remodeling processes.
4. Insulin-Related Metabolism
Insulin resistance, which is characterized by a reduced sensitivity to insulin in regulating blood glucose levels, is the primary pathological feature of type 2 diabetes mellitus. The role of ROS in insulin sensitivity is complex, with a dual effect: promoting insulin sensitivity in the early stages of disease, and contributing to insulin resistance as hyperglycemia progresses. The transient and controlled ROS production by NOXs in response to insulin is likely to be beneficial, while the chronic ROS generation by mitochondria during the context of prolonged nutrient overload in the later stages of the disease might be detrimental to insulin responsiveness [123,124]. Insulin stimulation can lead to this temporary increase in ROS levels by activating NOX and subsequently triggering insulin-mediated AKT activation. PIP3 and NOXs are located in the cell’s plasma membrane, suggesting that upon insulin stimulation, PTEN is oxidatively inactivated in close proximity to NOXs, and recruited PI3K can elevate PIP3 levels [125]. PIP3, in turn, triggers the PDK/AKT pathway, which subsequently phosphorylates various targets such as AS160, performing the anabolic effects of insulin stimulation [126,127]. The activated AKT pathway can enhance glucose absorption in adipocytes by facilitating the translocation of glucose transporter GLUT4 to the plasma membrane, as well as elevating GLUT1 expression. This aligns with the proposition that AKT signaling potentially participates in mediating insulin-stimulated responses [128]. Hence, as a negative regulator of the AKT pathway, the knockout of PTEN was experimentally shown to incrementally affect the level of GLUT4 expression in skeletal muscle and white adipose tissue, which consequently increases glucose uptake [129,130]. Additionally, in some studies, inhibiting PTEN’s PIP3-phosphatase activity has been proposed as a potential therapeutic approach for type 2 diabetes [131,132,133]. Loh et al. demonstrated that a slight increase in physiological ROS levels in muscle cells can induce PTEN oxidation and eventually enhance insulin-induced glucose uptake via the PI3K/AKT pathway [124]. Therefore, the redox regulation of PTEN holds promise as a method for managing type 2 diabetes mellitus (Figure 4).
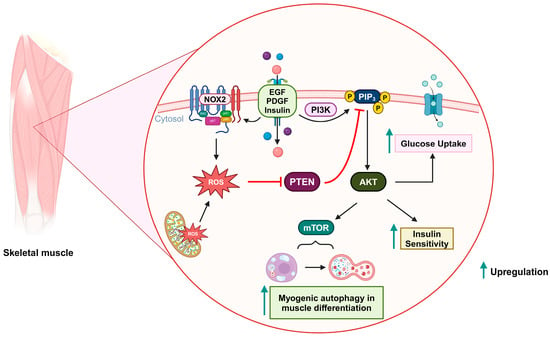
Figure 4. Oxidative inactivation of PTEN in insulin-related metabolism and muscle differentiation. Stimulation of growth factor receptors induces NOX2 activity and the production of ROS, which can oxidize PTEN and upregulate the PI3K/AKT signaling pathway. As a result, glucose uptake and insulin sensitivity are increased. During muscle differentiation, mitochondria-derived ROS can also oxidize PTEN and promote mTOR-induced myogenic autophagy.
5. Myogenic Autophagy in Muscle Differentiation
Autophagy is a crucial intracellular recycling process that eliminates old and dysfunctional cellular proteins and organelles. This process involves the formation of autophagosomes, which envelop parts of the cell’s cytoplasm that contain unnecessary components. As a result, autophagy functions as a dynamic mechanism for maintaining cellular health and resource efficiency [134,135]. Kim et al. demonstrated that the PI3K/AKT/mTOR signaling pathway is upregulated by mitochondrial ROS-derived H2O2, which subsequently implicates myogenesis-specific autophagy during muscle differentiation. In this scenario, PTEN is inactivated via oxidation [136] (Figure 4).
This entry is adapted from the peer-reviewed paper 10.3390/antiox13020199
This entry is offline, you can click here to edit this entry!