1. Mechanism of Angiogenesis in Tumors and Immune Checkpoints
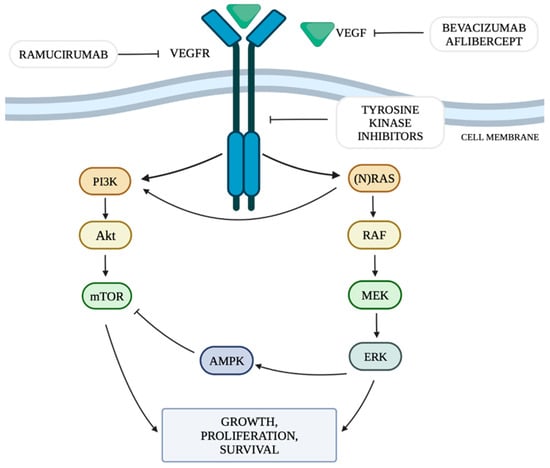
The mechanism of neoangiogenesis is largely dependent on the concentration of VEGF. It can activate cellular pathways that regulate cell division, migration, survival and apoptosis. These pathways include the PI3K/Akt signaling pathway (the phosphatidylinositol 3-kinase and serine-threonine protein kinase Akt pathway) and RAS/MAPK/ERK (rat sarcoma protooncogene/mitogen-activated protein kinase/extracellular signal-regulated kinase) by mutation or constitutive activation of their components [
14].
The VEGFR family includes five types of receptors, of which VEGFR-1 and VEGFR-2 play a role in angiogenesis. The binding of VEGF to VEGFR on the tumor blood vessel increases vascular permeability and activates the proliferation and migration of vascular endothelial cells [
4]. It has been shown that increased VEGF concentration results in “upregulation” of the VEGFR-2 receptor [
15]. The main factor stimulating VEGF is hypoxia. Chronic oxygen deficiency in cells increases the production of hypoxia-inducible factor 1 (HIF-1), which, in turn, is a transcription factor for increasing the production and release of VEGF [
16]. The increase in VEGF activation affects the activation of pathways responsible for neoangiogenesis in vascular endothelial cells, which is intended to counteract the effects of hypoxia. This increases the efficiency of endothelial cell proliferation, growth and migration processes and vascular permeability [
16]. The VEGF family includes: VEGFA, VEGFB, VEGFC, VEGFD and PlGF (placental growth factor). Signaling by VEGFA is mainly associated with the stimulation of VEGFR-2 [
17]. VEGF can be produced by tumor cells, as well as endothelial and stromal cells. Immune system cells also have the ability to produce VEGF [
3]. Moreover, cytokines such as VEGF that enter the systemic circulation from the TME may also enhance the extravasation of metastatic tumor cells from blood vessels in distant organs [
3]. Circulating VEGF and the soluble form of VEGFR-2 can be used as a predictor of response to antiangiogenic treatment [
18].
The RAS/MAPK/ERK pathway is responsible for transmitting signals from the cell surface to its nucleus, controlling the processes of proliferation, differentiation, migration and survival. The combination of a ligand with a receptor tyrosine kinase (RTK) protein results in the phosphorylation of the RAS (rat sarcoma) protein, which in turn activates the RAF (rapidly accelerated fibrosarcoma) protein with serine-threonine kinase activity. The RAF protein has three isoforms: ARAF, BRAF and CRAF, among which BRAF is the strongest activator of MEK (mitogen-activated kinase). The activated RAF kinase phosphorylates MEK1 and MEK2, which then activate the ERK1 and ERK2 kinases (extracellular signal-regulated kinases). Activated ERK kinases transmit a signal to the cell nucleus, which results in increased expression of genes responsible for cell growth and survival [
19]. Among all RTKs, a special role of the vascular endothelial growth factor receptor (VEGFR) and its ligands is indicated in the pathogenesis of cancer [
20]. The presence of KRAS mutations is expected to increase VEGF activity, thus promoting the process of tumor neoangiogenesis [
21].
The phosphatidylinositol 3-kinase and serine-threonine protein kinase Akt (PI3K/Akt) signaling pathway is one of the main metabolic pathways whose disturbances are observed in numerous cancers. They are related to the increased expression of genes whose products are involved in the PI3K/Akt pathway [
22]. PI3K is regulated mainly by RTK. The PI3K family includes three classes of kinases, of which class I is the best known and the most important in the context of oncogenesis and pathogenesis of other diseases [
23]. In class I PI3K, we can distinguish two subgroups: IA, which includes the α, β and δ isoforms and IB, which includes the γ isoform [
23]. PI3Kα plays a key role in cellular pathways related to angiogenesis, growth and metabolism, being the isoform most involved in the process of oncogenesis. Ligands that activate the pathway by attaching to RTKs include VEGF, among others. The activation of PI3K leads to an increase in the production of phosphatidylinositol 3,4,5-trisphosphate (PIP3) by phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2). PIP3 is responsible for increasing recruitment to the cell membrane and activation of serine/threonine kinase Akt, also called protein kinase B (PKB). The Akt kinase family includes three proteins: Akt-1, Akt-2 and Akt-3. Activated Akt kinase regulates, among others, the activity of a pro-apoptotic protein located on the outer mitochondrial membrane from the Bcl-2 family—the BAD protein (Bcl-2-associated death promoter)—and the serine-threonine kinase mTOR (serine/threonine kinase mammalian target of rapamycin), thus regulating the processes of apoptosis, angiogenesis and proliferation. The mTOR kinase functions as a catalytic subunit in two different protein complexes, mTORC1 and mTORC2 (mTOR complex 1,2). The PI3K/Akt pathway is controlled by the PTEN phosphatase encoded by the tumor suppressor gene PTEN. PTEN phosphatase is responsible for the conversion of PIP3 to PIP2 by catalyzing the separation of the 3′-phosphate group from PIP3, thus acting as a negative regulator of PI3K. When all components of the pathway function properly, they stop the proliferation process and initiate apoptosis [
23].
Figure 1 shows the discussed cellular pathways and the site of action of selected anti-VEGF drugs.
Figure 1. RAS/MAPK/ERK and PI3K/Akt/mTOR pathway and targets for anti-VEGF therapy. Own study based on [
6,
7,
8,
19,
22].
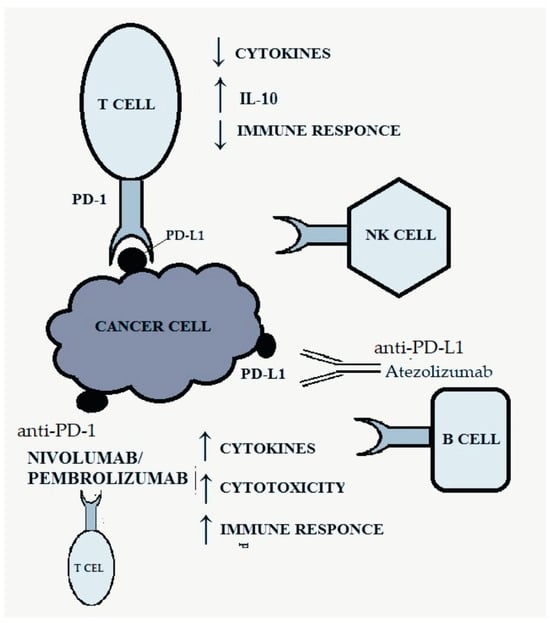
The PD-1 receptor, belonging to the CD28 family, is present on B lymphocytes, CD4+ and CD8+ T lymphocytes, natural killer (NK) cells, monocytes and activated dendritic cells and is responsible for inhibiting the anti-tumor response [
19,
24]. It has two ligands: PD-L1 and PD-L2 (programmed death-ligand 1, 2), the first of which is much more widespread and occurs on many different tissues [
25]. The combination of the ligand with the PD-1 receptor results in inhibition of the activity of the immune system by reducing the production of cytokines and increasing the synthesis of IL-10, which inhibits the immune response [
8,
19]. Activation of the PD-1-related pathway allows cancer cells to evade the body’s defense response by negatively regulating effector T cells [
8,
19].
Figure 2 shows the discussed pathway and the site of action of anti-PD-1/PD-L1 drugs.
Figure 2. PD-1/PD-L1 pathway and targets for anti-PD-1/PD-L1 therapy. Own study based on [
8,
24,
25].
2. Cardiotoxicity Associated with the Use of Anti-VEGF Therapy
The use of vascular endothelial growth factor inhibitors (VEGFI) and TKIs in oncological therapy may lead to cardiotoxicity expressed by arterial hypertension, left ventricular systolic dysfunction, heart failure and arterial and venous thromboembolism, as well as prolongation of the QTc interval and arrhythmia [
26]. The most frequently observed adverse effect associated with the use of VEGFI in relation to the cardiovascular system is hypertension. It is estimated that it affects up to 80–90% of patients treated with such therapy [
27]. In a large meta-analysis of 77 clinical trials of VEGF signaling pathway (VSP) inhibitors, severe hypertension occurred in 7.4% of patients, arterial thromboembolism in 1.8%, myocardial ischemia in 1.7% and myocardial failure in 2.3%. The use of VSP inhibitors was associated with a 5.6-fold increase in the risk of severe hypertension, a 2.8-fold increase in the risk of myocardial ischemia, a 1.5-fold increase in the risk of arterial thromboembolism and a 1.4-fold increase in the risk of myocardial failure [
28]. The development of hypertension contributes to the dilatation of the ascending aorta, hypertrophy of the interventricular septum and hypertrophy of the left ventricular muscle, which are visible on cardiac ultrasound [
29]. Changes typical of this type of myocardial remodeling, characteristic of left ventricular hypertrophy, can be observed on cardiac ultrasound [
30]. Studies have shown that EKG is not a reliable way to assess cardiac hypertrophy [
31,
32]. Echocardiography is still the only reliable assessment of cardiac hypertrophy [
31,
32].
Meta-analyses of data from randomized clinical trials showed that the use of TKIs, such as sunitinib, sorafenib or pazopanib, was associated with an increased risk of cardiac complications, and due to the lower specificity of the action of TKIs than anti-VEGF antibodies, they may cause even greater damage [
10]. The most frequently observed events were hypertension, arterial thrombotic events and decreased left ventricular ejection fraction. Interestingly, there was no increase in the risk of venous thrombotic events associated with their use [
10]. In one of the studies available in the literature, 829 patients with renal cell carcinoma treated with TKIs (pazopanib, sunitinib or sorafenib) and 2601 patients treated with bevacizumab were analyzed. During the one-year follow-up, in the group of patients treated with TKIs, 81 cases of major adverse cardiovascular events (MACEs) were recorded (CCI: 9.79%), of which 33 cases were thromboembolic events (CCI: 3.99%), 22 cases cardiac arrhythmia (CCI: 2.66%), 13 pulmonary embolism (CCI: 1.57%) and 13 heart failure (CCI: 1.57%). The incidence of adverse cardiovascular events was higher in the group of patients treated with TKIs than in the group of patients treated with bevacizumab, where a total of 176 events were recorded (CCI: 6.77%), with a predominance of thromboembolic complications (70 cases, CCI: 2.69%). The frequency of MACEs increased with the duration of therapy, both in the group of patients treated with TKIs (4.34% after 3 months, 9.79% after 1 year) and bevacizumab (2.38% after 3 months, 6.77% after 1 year). Interestingly, the incidence of arrhythmias, pulmonary embolism and heart failure associated with TKI use plateaued after 6 months of therapy, while the incidence of thromboembolic events continued to increase. The risk group included patients >65 years of age [
33]. During sunitinib treatment, changes in blood pressure were most pronounced during the first three cycles, and the greatest increases in the incidence of grade 3 hypertension and grade 3 cardiac dysfunction were observed in cycles 2 and 3 [
10].
The actual incidence of left ventricular systolic dysfunction (LVSD) associated with VEGFI may range from 10 to 20%, and the incidence of heart failure is approximately 1–5% [
26].
The incidence of heart failure in the case of bevacizumab was 2–4%, while in the case of TKI use it was 3–8%, and diabetics were identified as a group at particular risk of myocardial blood supply disorders, probably due to progressive myocardial fibrosis and deterioration of heart muscle function associated with diabetes [
10]. Interestingly, the use of TKIs may result in a decrease in glucose levels, even leading to the possibility of discontinuing hypoglycemic drugs in diabetic patients [
34]. A meta-analysis of 3784 breast cancer patients treated with bevacizumab in randomized, controlled trials found an incidence of “high-grade” congestive heart failure of 1.6% with bevacizumab and 0.4% with placebo. The term “high grade” refers to heart failure grade 3 or higher according to the National Cancer Institute’s (NCI) Common Terminology Criteria for Adverse Events, which includes patients with LVEF < 40% and a spectrum of clinical symptoms from patients with mild symptoms to cardiogenic shock and death [
35]. Although the incidence was low, the relative risk (RR) of developing symptomatic heart failure was 4.74 when bevacizumab was added to anticancer treatment regimens, compared with the addition of placebo [
36]. A meta-analysis of studies including a total of 10,647 patients treated with TKIs for various cancers (including pazopanib, sorafenib and sunitinib) showed an overall incidence of asymptomatic LVSD of 2.4%, with 1.2% developing symptomatic heart failure [
37]. A larger meta-analysis of 28,538 patients showed similar results. TKIs were associated with the highest RR for “high-grade” cardiotoxicity of 5.62, compared with placebo-treated patients. Of note, ramucirumab and aflibercept were also among the drugs with the highest relative risk of “high-grade” cardiotoxicity—5.0 and 4.1, respectively [
38]. In a randomized trial of 1110 patients comparing pazopanib in combination with sunitinib for the treatment of renal cell carcinoma, the incidence of heart failure was 1%, while 9% of patients experienced a ≥15% reduction in LVEF [
39]. The incidence of myocardial ischemia in patients treated with anti-VEGF antibodies is relatively low. In a meta-analysis of 4617 patients, bevacizumab-treated patients had a significantly increased risk of coronary heart disease (defined as myocardial infarction, unstable angina, coronary revascularization, coronary artery disease, arrhythmias, sudden death or cardiovascular death), compared with control (RR: 2.49). However, the overall incidence of coronary heart disease was only 1.0% [
40]. In another meta-analysis of 72 clinical trials and 38,078 patients treated with VEGFI, 4136 cases of myocardial infarction were reported. VEGFI had a significantly increased relative risk of myocardial infarction (RR: 3.54) compared to the control group, although the absolute risk was only 0.8% [
41].
Cardiac arrhythmias and prolongation of the QTc interval may also occur during anti-VEGF therapy, but the frequency and degree of increased risk of cardiac disorders associated with such therapy have not yet been specified. The TKIs most frequently associated with QTc prolongation are sorafenib and sunitinib. There are no reports of QTc prolongation with bevacizumab in clinical trials [
26], and a study of 87 cancer patients treated with aflibercept, which has a higher affinity for binding VEGF, showed a mean increase in the QTc interval of only 8.4 ms [
42]. Atrial fibrillation (AF) was most commonly reported following sorafenib administration. The incidence of AF was 5.1% when used in combination with 5-fluorouracil in a study of 39 patients with advanced hepatocellular carcinoma [
43].
3. Mechanism of Cardiotoxicity Associated with the Use of Anti-VEGF Therapy
Cardiac muscle cells express all VEGF isoforms and VEGF receptor subtypes. This axis is stimulated by hypoxia and ischemia, as well as stretching of the myocardium. Cardiomyocytes release VEGF upon exposure to various stressors, which then act on vascular endothelial cells in a paracrine manner. Vascular endothelial cells also have the ability to release VEGF [
10]. VEGF will contribute to cardiomyocyte turnover by activating mitogen-activated protein kinase (MAPK) pathways, which play a key regulatory role in cell proliferation in cardiomyocytes. The mitogenic effect of VEGF on endothelial cells is mediated by VEGFR-2 and activation of extracellular signal-regulated kinase (ERK1/2). The consequence of VEGFR-2 phosphorylation is the stimulation of the RAS/RAF/MAPK/ERK pathway in endothelial cells. In addition to its effects on VEGFR, sorafenib is a potent RAF inhibitor. VEGF also plays a role in cell migration, including endothelial cells. Cell migration is induced, among others, by the PI3K/Akt and MAPK pathways [
26].
The role of AMPK (5′AMP-activated protein kinase), which is crucial for the energy homeostasis of cardiomyocytes, is also important. A decrease in ATP levels leads to the activation of AMPK and inhibition of protein and fatty acid synthesis. VEGF inhibitors can reduce the level of AMPK, which reduces its activation in response to ATP deficiency and, at the same time, affects the activation of mTOR [
26].
Coronary vasoconstriction is regulated by the concentration of nitric oxide (NO). VEGF, by influencing VEGFR-2, causes its endothelial release. As a result of the use of VEGF inhibitors, the bioavailability of NO is reduced, which results in vasoconstriction, and is potentially the most important factor responsible for the occurrence of hypertension. Endothelin-1 (ET-1), responsible for vasoconstriction, may also play a role in this process. It has been shown that NO suppression and an increase in ET-1 concentration occurred in patients treated with regorafenib [
26].
Moreover, NO, like prostacyclin (PGI2), has antiplatelet effects. Therefore, VEGFI-induced suppression of endothelial NO and PGI2 will result in increased coagulation. This increases the risk of arterial thrombosis and venous thromboembolism [
26]. It has been shown that simultaneous administration of heparin with bevacizumab increases the deposition of bevacizumab-VEGF immune complexes on platelets, which leads to their activation [
26].
Decreased production of nitric oxide and the resulting vasoconstriction may also contribute to the developing endothelial dysfunction, which promotes inflammation, atherosclerosis and platelet reactivity. Coronary vasoconstriction superimposed on structural changes may lead to a significant reduction in perfusion pressure and myocardial ischemia and failure [
10].
One of the potential mechanisms of VEGFI-induced cardiac arrhythmias may be the inhibition of the PI3K/Akt pathway in cardiomyocytes. Binding of VEGF to VEGFR-2 on endothelial cells is associated with increased cell survival and migration through activation of the PI3K/Akt pathway. Inhibition of VEGF-induced signaling leads to a decrease in PI3K activity, among others, in the atrial appendage, leading to the occurrence of AF, which was proven in mouse models [
26].
4. Cardiotoxicity Associated with the Use of Anti-PD-1/PD-L1 Therapy
The most frequently diagnosed cardiovascular irAE (immune-related adverse event) is myocarditis (79% of all cases), but arrhythmias may also occur, including atrial fibrillation (30%), conduction disturbances (17%) or ventricular arrhythmias (27%), pericardial diseases, vasculitis or takotsubo-like cardiomyopathy (14%) [
12]. The incidence of myocarditis induced by anti-PD-1/PD-L1 therapy ranges from 0.09 to 2% but is associated with high mortality—up to half of cases. The onset of myocarditis associated with immunotherapy most often occurs at an early stage of treatment, with up to 80% occurring within the first 3 months after the initiation of therapy, i.e., most often after 2–3 treatment cycles [
12]. Cardiac complications mostly lead to the termination of anti-PD-1/PD-L1 treatment.
Activated immune cells can attack not only myocardial but also pericardial antigens, which may lead to pericarditis with pericardial effusion or even cardiac tamponade. Pericarditis is a less common adverse event compared to myocarditis, with an incidence of up to 0.3%. Another adverse effect is vasculitis (with incidence estimated at approximately 0.26% [
44]), most often polymyalgia rheumatica and temporal arteritis. The mortality rate for these complications is lower compared to other cardiovascular irAEs (6.1%), and visual disturbances were reported in 27.8% of cases of temporal arteritis [
12].
Acute coronary syndrome associated with immunotherapy has an incidence of approximately 1%. A single-center study of 3326 patients undergoing immunotherapy showed a 7% incidence of myocardial infarction and a similar incidence of stroke over a mean follow-up period of 16 months [
45].
5. Mechanism of Cardiotoxicity Associated with the Use of Anti-PD-1/PD-L1 Therapy
Cardiac complications also occur in the case of immunotherapy using PD-1/PD-L1 inhibitors. The PD-1/PD-L1 pathway seems to be crucial for immune homeostasis within the myocardium and for protection of the heart against T lymphocytes. The mechanism of cardiotoxicity induced by PD-1/PD-L1 inhibitors is not fully understood. One of the causes is immunological dysregulation in the heart muscle caused by excessive activation of native T lymphocytes. Another cause may be cross-reactions between T lymphocytes and antigens present in cardiac muscle cells. Dysregulated immune cells can falsely label surface structures such as cardiolipin as antigens. As a result, normal cardiomyocytes expressing such an antigen may become a target of the immune system. The third possible mechanism is a systemic immune response triggering the release of cytokines [
12,
44].
This entry is adapted from the peer-reviewed paper 10.3390/genes15020177