Apoptosis, or programmed cell death, is a fundamental process that maintains tissue homeostasis, eliminates damaged or infected cells, and plays a crucial role in various biological phenomena. The deregulation of apoptosis is involved in many human diseases, including cancer. One of the emerging players in the intricate regulatory network of apoptosis is apoptosis inhibitor 5 (API5), also called AAC-11 (anti-apoptosis clone 11) or FIF (fibroblast growth factor-2 interacting factor). While it may not have yet the same level of notoriety as some other cancer-associated proteins, API5 has garnered increasing attention in the cancer field, as elevated API5 levels are often associated with aggressive tumor behavior, resistance to therapy, and poor patient prognosis.
- apoptosis inhibitor 5
- apoptosis
- cancer
1. Introduction
2. Apoptosis Inhibitor 5 and Cancer
2.1. API5 Expression and Prognosis Value
2.2. API5’s Role on Cancer Metastasis, Immune Response, and Survival


2.3. Targeting API5 as a Therapeutic Approach
This entry is adapted from the peer-reviewed paper 10.3390/biom14010136
References
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. Apoptosis: Mechanisms and relevance in cancer. Ann. Hematol. 2005, 84, 627–639.
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344.
- Geske, F.J.; Gerschenson, L.E. The biology of apoptosis. Hum. Pathol. 2001, 32, 1029–1038.
- Meier, P.; Vousden, K.H. Lucifer’s labyrinth—Ten years of path finding in cell death. Mol. Cell 2007, 28, 746–754.
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288.
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87.
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448.
- Tuveson, D.; Hanahan, D. Translational medicine: Cancer lessons from mice to humans. Nature 2011, 471, 316–317.
- Green, D.R. Caspases and Their Substrates. Cold Spring Harb. Perspect. Biol. 2022, 14, a041012.
- Lamkanfi, M.; Festjens, N.; Declercq, W.; Berghe, T.V.; Vandenabeele, P. Caspases in cell survival, proliferation and differentiation. Cell Death Differ. 2007, 14, 44–55.
- Thornberry, N.A.; Lazebnik, Y. Caspases: Enemies within. Science 1998, 281, 1312–1316.
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206.
- Shi, Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 2002, 9, 459–470.
- Krueger, A.; Schmitz, I.; Baumann, S.; Krammer, P.H.; Kirchhoff, S. Cellular FLICE-inhibitory protein splice variants inhibit different steps of caspase-8 activation at the CD95 death-inducing signaling complex. J. Biol. Chem. 2001, 276, 20633–20640.
- Wang, Z.; Figueiredo-Pereira, C.; Oudot, C.; Vieira, H.L.A.; Brenner, C. Mitochondrion: A Common Organelle for Distinct Cell Deaths? Int. Rev. Cell Mol. Biol. 2017, 331, 245–287.
- Bao, Q.; Shi, Y. Apoptosome: A platform for the activation of initiator caspases. Cell Death Differ. 2007, 14, 56–65.
- Levine, B.; Sinha, S.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606.
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-suppressor Genes in Carcinogenesis: A Review. Anticancer. Res. 2020, 40, 6009–6015.
- Plati, J.; Bucur, O.; Khosravi-Far, R. Apoptotic cell signaling in cancer progression and therapy. Integr. Biol. 2011, 3, 279–296.
- Reed, J.C. Apoptosis-targeted therapies for cancer. Cancer Cell 2003, 3, 17–22.
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208.
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.-N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482.
- Caenepeel, S.; Brown, S.P.; Belmontes, B.; Moody, G.; Keegan, K.S.; Chui, D.; Whittington, D.A.; Huang, X.; Poppe, L.; Cheng, A.C.; et al. AMG 176, a Selective MCL1 Inhibitor, Is Effective in Hematologic Cancer Models Alone and in Combination with Established Therapies. Cancer Discov. 2018, 8, 1582–1597.
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341.
- Tewari, M.; Yu, M.; Ross, B.; Dean, C.; Giordano, A.; Rubin, R. AAC-11, a novel cDNA that inhibits apoptosis after growth factor withdrawal. Cancer Res. 1997, 57, 4063–4069.
- Basset, C.; Bonnet-Magnaval, F.; Navarro, M.G.-J.; Touriol, C.; Courtade, M.; Prats, H.; Garmy-Susini, B.; Lacazette, E. Api5 a new cofactor of estrogen receptor alpha involved in breast cancer outcome. Oncotarget 2017, 8, 52511–52526.
- Bousquet, G.; Feugeas, J.-P.; Gu, Y.; Leboeuf, C.; El Bouchtaoui, M.; Lu, H.; Espié, M.; Janin, A.; Di Benedetto, M. High expression of apoptosis protein (Api-5) in chemoresistant triple-negative breast cancers: An innovative target. Oncotarget 2019, 10, 6577–6588.
- Cho, H.; Chung, J.-Y.; Song, K.-H.; Noh, K.H.; Kim, B.W.; Chung, E.J.; Ylaya, K.; Kim, J.H.; Kim, T.W.; Hewitt, S.M.; et al. Apoptosis inhibitor-5 overexpression is associated with tumor progression and poor prognosis in patients with cervical cancer. BMC Cancer 2014, 14, 545.
- Kim, J.W.; Cho, H.S.; Kim, J.H.; Hur, S.Y.; Kim, T.E.; Lee, J.M.; Kim, I.K.; Namkoong, S.E. AAC-11 overexpression induces invasion and protects cervical cancer cells from apoptosis. Lab. Investig. 2000, 80, 587–594.
- Krejci, P.; Pejchalova, K.; Rosenbloom, B.E.; Rosenfelt, F.P.; Tran, E.L.; Laurell, H.; Wilcox, W.R. The antiapoptotic protein Api5 and its partner, high molecular weight FGF2, are up-regulated in B cell chronic lymphoid leukemia. J. Leukoc. Biol. 2007, 82, 1363–1364.
- Noh, K.H.; Kim, S.-H.; Kim, J.H.; Song, K.-H.; Lee, Y.-H.; Kang, T.H.; Han, H.D.; Sood, A.K.; Ng, J.; Kim, K.; et al. API5 confers tumoral immune escape through FGF2-dependent cell survival pathway. Cancer Res. 2014, 74, 3556–3566.
- Sasaki, H.; Moriyama, S.; Yukiue, H.; Kobayashi, Y.; Nakashima, Y.; Kaji, M.; Fukai, I.; Kiriyama, M.; Yamakawa, Y.; Fujii, Y. Expression of the antiapoptosis gene, AAC-11, as a prognosis marker in non-small cell lung cancer. Lung Cancer 2001, 34, 53–57.
- Song, K.H.; Cho, H.; Lee, H.J.; Oh, S.J.; Woo, S.R.; Hong, S.O.; Jang, H.S.; Noh, K.H.; Choi, C.H.; Chung, J.Y.; et al. API5 confers cancer stem cell-like properties through the FGF2-NANOG axis. Oncogenesis 2017, 6, e285.
- Song, K.H.; Kim, S.H.; Noh, K.H.; Bae, H.C.; Kim, J.H.; Lee, H.J.; Song, J.; Kang, T.H.; Kim, D.W.; Oh, S.J.; et al. Apoptosis Inhibitor 5 Increases Metastasis via Erk-mediated MMP expression. BMB Rep. 2015, 48, 330–335.
- Wang, Z.; Liu, H.; Liu, B.; Ma, W.; Xue, X.; Chen, J.; Zhou, Q. Gene expression levels of CSNK1A1 and AAC-11, but not NME1, in tumor tissues as prognostic factors in NSCLC patients. Med. Sci. Monit. 2010, 16, CR357-64.
- Carvalho, R.; Milne, A.N.; Polak, M.; Offerhaus, G.J.; Weterman, M.A. A novel region of amplification at 11p12-13 in gastric cancer, revealed by representational difference analysis, is associated with overexpression of CD44v6, especially in early-onset gastric carcinomas. Genes Chromosomes Cancer 2006, 45, 967–975.
- Klingbeil, P.; Natrajan, R.; Everitt, G.; Vatcheva, R.; Marchio, C.; Palacios, J.; Buerger, H.; Reis-Filho, J.S.; Isacke, C.M. CD44 is overexpressed in basal-like breast cancers but is not a driver of 11p13 amplification. Breast Cancer Res. Treat. 2010, 120, 95–109.
- Jarvinen, A.K.; Autio, R.; Kilpinen, S.; Saarela, M.; Leivo, I.; Grénman, R.; Mäkitie, A.A.; Monni, O. High-resolution copy number and gene expression microarray analyses of head and neck squamous cell carcinoma cell lines of tongue and larynx. Genes Chromosomes Cancer 2008, 47, 500–509.
- Fukuda, Y.; Kurihara, N.; Imoto, I.; Yasui, K.; Yoshida, M.; Yanagihara, K.; Park, J.-G.; Nakamura, Y.; Inazawa, J. CD44 is a potential target of amplification within the 11p13 amplicon detected in gastric cancer cell lines. Genes Chromosomes Cancer 2000, 29, 315–324.
- Kuttanamkuzhi, A.; Panda, D.; Malaviya, R.; Gaidhani, G.; Lahiri, M. Altered expression of anti-apoptotic protein Api5 affects breast tumorigenesis. BMC Cancer 2023, 23, 374.
- Krejci, P.; Koci, L.; Chlebova, K.; Hyzdalova, M.; Hofmanova, J.; Jira, M.; Kysela, P.; Kozubik, A.; Kala, Z. Apoptosis inhibitor 5 (API-5; AAC-11; FIF) is upregulated in human carcinomas in vivo. Oncol. Lett. 2012, 3, 913–916.
- Ren, K.; Zhang, W.; Shi, Y.; Gong, J. Pim-2 activates API-5 to inhibit the apoptosis of hepatocellular carcinoma cells through NF-kappaB pathway. Pathol. Oncol. Res. 2010, 16, 229–237.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Curran, S.; Murray, G.I. Matrix metalloproteinases: Molecular aspects of their roles in tumour invasion and metastasis. Eur. J. Cancer 2000, 36, 1621–1630.
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 2002, 295, 2387–2392.
- Wu, B.; Crampton, S.P.; Hughes, C.C. Wnt signaling induces matrix metalloproteinase expression and regulates T cell transmigration. Immunity 2007, 26, 227–239.
- Costa, D.B.; Halmos, B.; Kumar, A.; Schumer, S.T.; Huberman, M.S.; Boggon, T.J.; Tenen, D.G.; Kobayashi, S. BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations. PLoS Med. 2007, 4, 1669–1679, discussion 1680.
- Hubner, A.; Barrett, T.; Flavell, R.A.; Davis, R.J. Multisite phosphorylation regulates Bim stability and apoptotic activity. Mol. Cell 2008, 30, 415–425.
- Morris, E.J.; Michaud, W.A.; Ji, J.-Y.; Moon, N.-S.; Rocco, J.W.; Dyson, N.J. Functional identification of Api5 as a suppressor of E2F-dependent apoptosis in vivo. PLoS Genet. 2006, 2, e196.
- Rigou, P.; Piddubnyak, V.; Faye, A.; Rain, J.-C.; Michel, L.; Calvo, F.; Poyet, J.-L. The antiapoptotic protein AAC-11 interacts with and regulates Acinus-mediated DNA fragmentation. EMBO J. 2009, 28, 1576–1588.
- Janin, Y.L. Peptides with anticancer use or potential. Amino Acids 2003, 25, 1–40.
- Imre, G.; Berthelet, J.; Heering, J.; Kehrloesser, S.; Melzer, I.M.; Lee, B.I.; Thiede, B.; Dötsch, V.; Rajalingam, K. Apoptosis inhibitor 5 is an endogenous inhibitor of caspase-2. EMBO Rep. 2017, 18, 733–744.
- Chen, M.; Wang, L.; Li, M.; Budai, M.M.; Wang, J. Mitochondrion-Mediated Cell Death through Erk1-Alox5 Independent of Caspase-9 Signaling. Cells 2022, 11, 3053.
- Chen, Q.; Song, S.; Wei, S.; Liu, B.; Honjo, S.; Scott, A.; Jin, J.; Ma, L.; Zhu, H.; Skinner, H.D.; et al. ABT-263 induces apoptosis and synergizes with chemotherapy by targeting stemness pathways in esophageal cancer. Oncotarget 2015, 6, 25883–25896.
- Ho, C.J.; Ko, H.-J.; Liao, T.-S.; Zheng, X.-R.; Chou, P.-H.; Wang, L.-T.; Lin, R.-W.; Chen, C.-H.; Wang, C. Severe cellular stress activates apoptosis independently of p53 in osteosarcoma. Cell Death Discov. 2021, 7, 275.
- Ahel, D.; Hořejší, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P.; et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 2009, 325, 1240–1243.
- Wang, Y.; Lee, A.T.C.; Ma, J.Z.I.; Wang, J.; Ren, J.; Yang, Y.; Tantoso, E.; Li, K.-B.; Ooi, L.L.P.J.; Tan, P.; et al. Profiling microRNA expression in hepatocellular carcinoma reveals microRNA-224 up-regulation and apoptosis inhibitor-5 as a microRNA-224-specific target. J. Biol. Chem. 2008, 283, 13205–13215.
- Chopra, M.; Dharmarajan, A.M.; Meiss, G.; Schrenk, D. Inhibition of UV-C light-induced apoptosis in liver cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 2009, 111, 49–63.
- Yuan, J.; Liu, Z.; Liu, J.; Fan, R. Circ_0060055 Promotes the Growth, Invasion, and Radioresistance of Glioblastoma by Targeting MiR-197-3p/API5 Axis. Neurotox. Res. 2022, 40, 1292–1303.
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70.
- Burnett, J.C.; Rossi, J.J. RNA-based therapeutics: Current progress and future prospects. Chem. Biol. 2012, 19, 60–71.
- Feng, R.; Patil, S.; Zhao, X.; Miao, Z.; Qian, A. RNA Therapeutics—Research and Clinical Advancements. Front. Mol. Biosci. 2021, 8, 710738.
- Sharma, V.K.; Lahiri, M. Interplay between p300 and HDAC1 regulate acetylation and stability of Api5 to regulate cell proliferation. Sci. Rep. 2021, 11, 16427.
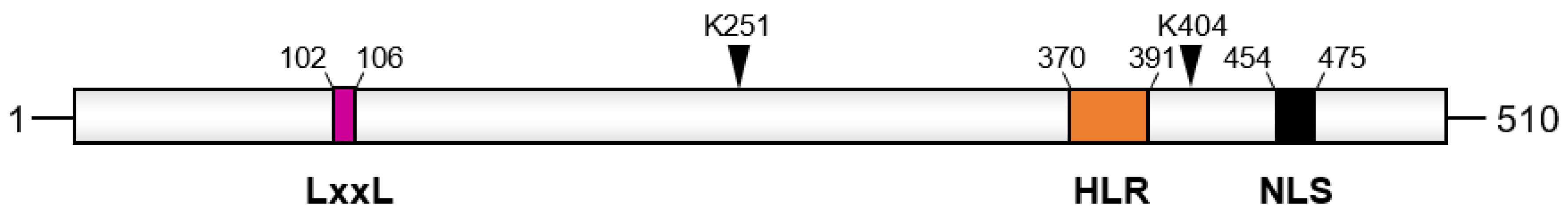
- Han, B.G.; Kim, K.H.; Lee, S.J.; Jeong, K.C.; Cho, J.W.; Noh, K.H.; Kim, T.W.; Kim, S.J.; Yoon, H.J.; Suh, S.W.; et al. Helical repeat structure of apoptosis inhibitor 5 reveals protein-protein interaction modules. J. Biol. Chem. 2012, 287, 10727–10737.
- Attar, N.; Kurdistani, S.K. Exploitation of EP300 and CREBBP Lysine Acetyltransferases by Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026534.
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356.
- Van den Berghe, L.; Laurell, H.; Huez, I.; Zanibellato, C.; Prats, H.; Bugler, B. FIF , a nuclear putatively antiapoptotic factor, interacts specifically with FGF-2. Mol. Endocrinol. 2000, 14, 1709–1724.
- Habault, J.; Thonnart, N.; Pasquereau-Kotula, E.; Bagot, M.; Bensussan, A.; Villoutreix, B.O.; Marie-Cardine, A.; Poyet, J.L. PAK1-dependent anti-tumor effect of AAC-11-derived peptides on Sézary syndrome malignant CD4+ T lymphocytes. J. Investig. Dermatol. 2021, 141, 2261–2271.
- Habault, J.; Thonnart, N.; Ram-Wolff, C.; Bagot, M.; Bensussan, A.; Poyet, J.-L.; Marie-Cardine, A. Validation of AAC-11-Derived Peptide Anti-Tumor Activity in a Single Graft Sezary Patient-Derived Xenograft Mouse Model. Cells 2022, 11, 2933.
- Jagot-Lacoussiere, L.; Kotula, E.; Villoutreix, B.O.; Bruzzoni-Giovanelli, H.; Poyet, J.L. A Cell-Penetrating Peptide Targeting AAC-11 Specifically Induces Cancer Cells Death. Cancer Res. 2016, 76, 5479–5490.
- Habault, J.; Kaci, A.; Pasquereau-Kotula, E.; Fraser, C.; Chomienne, C.; Dombret, H.; Braun, T.; Pla, M.; Poyet, J.-L. Prophylactic and therapeutic antileukemic effects induced by the AAC-11-derived Peptide RT53. OncoImmunology 2020, 9, 1728871.
- Habault, J.; Fraser, C.; Pasquereau-Kotula, E.; Born-Bony, M.; Marie-Cardine, A.; Poyet, J.-L. Efficient Therapeutic Delivery by a Novel Cell-Penetrating Peptide Derived from Acinus. Cancers 2020, 12, 1858.
- Pasquereau-Kotula, E.; Habault, J.; Kroemer, G.; Poyet, J.-L. The anticancer peptide RT53 induces immunogenic cell death. PLoS ONE 2018, 13, e0201220.
- Papo, N.; Shai, Y. Host defense peptides as new weapons in cancer treatment. Cell. Mol. Life Sci. 2005, 62, 784–790.
- Bong, S.M.; Bae, S.-H.; Song, B.; Gwak, H.; Yang, S.-W.; Kim, S.; Nam, S.; Rajalingam, K.; Oh, S.J.; Kim, T.W.; et al. Regulation of mRNA export through API5 and nuclear FGF2 interaction. Nucleic Acids Res. 2020, 48, 6340–6352.