1. Introduction
About 100 years ago, Otto Warburg made an interesting observation: cancer cells when cultured in vitro under normal oxygen levels (i.e., 20% or 160 mmHg) as in physiological conditions or tumors growing in the body in vivo consume glucose much more than normal cells but convert this glucose predominantly into lactic acid, which is released into the culture medium
[1][2]. This was unexpected because lactic acid is the end product of glucose breakdown only under conditions of oxygen deficit (i.e., hypoxia), a pathway known as “anerobic glycolysis”. In contrast to this normal process, cancer cells convert glucose into lactic acid in the presence of oxygen, thus leading to the coining of the term “aerobic glycolysis” to describe the observation made by Warburg in cancer cells. Though interesting and unexpected, the importance of this observation was not recognized for several decades. A part of the reason for this was the discovery of oncogenes and tumor suppressor genes and the domination of the idea in the field of cancer biology that the protein products of these genes are the principal drivers of cancer growth. Surprisingly, however, investigations into the molecular targets of these oncogenes and tumor suppressor genes began to underscore the importance of metabolic pathways as the likely mediators of these genes in cancer growth. As a result, the interest in the field of cancer biology has shifted in the past couple of decades to cancer-cell-specific metabolism. Naturally, the starting point for this shift was the Warburg effect, which represents the first metabolic pathway to be discovered that is specific to cancer cells. What followed in subsequent years in this field is simply a logical extension of the original observation by Warburg.
The greater than normal consumption of glucose in cancer cells brought attention to glucose transporters, which deliver glucose into these cells. Since lactic acid is generated at high levels, cancer cells must find ways to prevent cellular acidification and get rid of lactic acid. This shifted the focus to lactate transporters that mediate the transfer of lactate and H
+ across the plasma membrane
[3][4]. Then came the discovery that lactate controls the proteasomal degradation of hypoxia-inducible factor-1α (HIF-1α), thus increasing HIF-1α protein levels in cancer cells even in the presence of normal oxygen, a condition now described as “pseudohypoxia”
[5]. Since cancer cells release massive amounts of lactic acid into the tumor microenvironment, which results in extracellular acidification, investigations began to explore the role of acidic pH in the tumor microenvironment in cancer growth. This led to the focus on H
+-coupled transporters for various nutrients such as amino acids, peptides, citrate, folate, and iron, which might use the extracellular acidic pH to effectively transfer these important nutrients to cancer cells from the extracellular medium
[6]. With the increased rate of glycolysis came increased levels of metabolic intermediates in the pathway. This brought attention to the use of these intermediates for the anabolic pathways to generate amino acids such as serine and glycine, which are obligatory as the source of one-carbon moieties for one-carbon metabolism involved in the synthesis of purines, pyrimidines, and thymidine monophosphate (TMP)
[7][8].
Cancer cells must have a greater need than normal cells for metabolic energy to support their high rate of proliferation, but the glucose–lactic acid pathway that occurs in these cells generates only a fraction of energy compared to the complete oxidation of glucose to CO
2 (2 ATP versus 32 ATP). This raised the possibility of other metabolic pathways generating ATP, which led to the discovery of the obligate dependence of cancer cells on extracellular glutamine (glutamine addiction). Subsequent research showed that glutamine is used in cancer cells not only for ATP generation but also for lactic acid production (glutaminolysis) and fatty acid synthesis (reductive carboxylation)
[9][10][11]. This also brought attention to amino acid transporters in the plasma membrane of cancer cells, which deliver not only glutamine to satisfy the “glutamine addiction” but also serine and glycine to fuel the one-carbon metabolism in addition to serine and glycine that are synthesized from the glycolytic intermediates
[12][13][14][15]. Cancer cells obtain amino acids not only from the extracellular medium but also from the lysosomal degradation of intracellular proteins via autophagy and extracellular proteins via macropinocytosis
[16][17]. This necessitated studies on amino acid transporters in the lysosomal membrane in cancer cells that transfer amino acids from lysosomes into cytoplasm for subsequent use in metabolic pathways
[18][19]. This also integrated amino acid nutrition to mTORC1 signaling because of the close association of mTORC1-associated proteins with the lysosomal membrane, a critical signaling pathway necessary for cancer cell proliferation and growth
[18][19].
Cancer cells are also addicted to iron and heme because of their essential role in a multitude of metabolic processes involved in energy production as well as catabolic and anabolic processes
[20][21]. Some of the critical steps in heme synthesis occur within the mitochondrial matrix, including the first regulatory step in the pathway, which combines glycine with succinyl-CoA to generate δ-aminolevulinate. In addition, one-carbon metabolism also participates in important biochemical processes within the mitochondrial matrix that require serine. This brings attention to transporters in the mitochondrial membrane that deliver serine and glycine from cytoplasm to the mitochondrial matrix
[22]. Furthermore, with the accumulation of excess iron in cancer cells comes the risk of oxidative stress and the iron-dependent cell death process (ferroptosis)
[23][24][25]. Therefore, cancer cells must enhance their antioxidant machinery to protect themselves from these detrimental processes. This led to the focus on the glutathione/glutathione peroxidase system and pentose phosphate pathway, which produce the reducing equivalent NADPH necessary for the antioxidant machinery
[26]. In addition, since cysteine is the rate-limiting amino acid for glutathione synthesis, studies began on amino acid transporters in the plasma membrane and lysosomal membrane that provide this critical amino acid to cancer cells to support the enhanced glutathione production
[27][28][29].
The original explanation for “aerobic glycolysis” thought by Warburg was that mitochondria are damaged in cancer cells and therefore the oxygen-dependent metabolism of pyruvate in mitochondria is impaired. This means that almost all the metabolic energy needed for the cancer cells comes from the conversion of glucose to lactic acid. This view has now been revised considerably based on glutamine metabolism within the mitochondria in cancer cells. The source of energy production simply shifts to a significant extent from glycolysis to glutaminolysis with intact mitochondrial function necessary for the latter process. To support their growth and proliferation, cancer cells use the intermediates in the enhanced glycolytic pathway to feed into other metabolic pathways instead for energy production.
2. Hypoxia and Anaerobic Glycolysis in Normal Cells
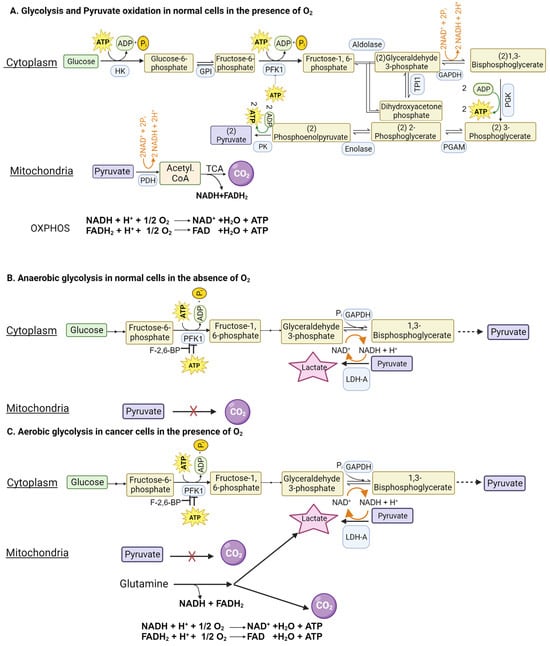
Glucose can be metabolized completely into CO2 and H2O in cells with mitochondria when oxygen is available. This complete oxidation of glucose involves glycolysis (glucose → pyruvate) in cytoplasm and the pyruvate dehydrogenase (PDH) and citric acid cycle (pyruvate → CO2) in the mitochondrial matrix (Figure 1A). However, neither glycolysis nor the PDH/citric acid cycle involves oxygen. When glucose goes through glycolysis and the PDH/citric acid cycle, it generates reducing equivalents NADH and FADH2, which enter the electron transport chain, and oxidative phosphorylation, where oxygen is used to convert these reducing equivalents back to NAD+ and FAD with the concomitant production of ATP. The entire process results in the generation of 32 ATP per glucose. This “aerobic glycolysis” associated with the complete oxidation of glucose in normal cells is subject to negative feedback regulation by ATP, which inhibits phosphofructokinase-1 (PFK-1), the most important rate-limiting enzyme in glycolysis. In other words, the aerobic glycolysis in normal cells is self-limiting, controlled by the energy status of the cell.
Figure 1. Glycolysis in normal cells and in cancer cells in the presence and absence of oxygen. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PFK1, phosphofructokinase-1; F-2,6-BP, fructose-2,6-bisphosphate.
When oxygen is deficient in normal cells, mitochondrial oxidation of pyruvate that is generated in glycolysis in the cytoplasm is impaired due to suppression of the electron transport chain (ETC) and oxidative phosphorylation (OXPHOS). Even though most of the NADH from glucose oxidation is produced within the mitochondria, the step mediated by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in glycolysis also generates NADH. If this NADH cannot be oxidized back to NAD+ due to defective ETC/OXPHOS as under hypoxic conditions, the reaction mediated by GAPDH cannot continue because of the lack of NAD+. This forces the conversion of pyruvate to lactate in the cytoplasm by lactate dehydrogenase (LDH), a reaction that substitutes for ETC/OXPHOS to convert NADH to NAD+ but without O2 consumption and ATP production (Figure 1B). Now, glycolysis can continue because of the functional coupling between GAPDH and LDH via NADH/NAD+ recycling, the entire process occurring in cytoplasm. This process where glucose gets converted to lactic acid in normal cells under hypoxic conditions is called “anaerobic glycolysis”. Interestingly, the same process occurs in erythrocytes even in the presence of oxygen because of the absence of mitochondria. Consequently, lactic acid is the end product of glycolysis in normal cells only under hypoxic conditions whereas erythrocytes generate lactic acid in glycolysis all the time.
3. Aerobic Glycolysis in Cancer Cells
3.1. Mechanisms used to Facilitate “Aerobic Glycolysis” in Cancer Cells
As mentioned above, glycolysis in normal cells in the presence of oxygen is self-limiting and pyruvate gets converted to CO
2. The conversion of glucose to lactic acid occurs in normal cells only under hypoxic conditions. In contrast, cancer cells metabolize glucose predominantly into lactic acid even in the presence of oxygen. The functional coupling between the reactions mediated by GAPDH and LDH is necessary for this process. Why does pyruvate not get oxidized to CO
2 in cancer cells when oxygen is available? This is mostly due to the defective transport of pyruvate from cytoplasm into the mitochondrial matrix because of the cancer-associated downregulation of the pyruvate-carrier components MPC1 and MPC2 in the inner mitochondrial membrane
[30] and the decreased catalytic activity of PDH due to an increased expression of PDH kinase-1 (PDK-1) and PDH kinase-3 (PDK-3) and consequent increased phosphorylation of PDH
[31]. An additional mechanism for the inactivation of PDH in cancer cells involves signaling pathways associated with EGFR activation and mutant K-Ras
[32]. In this mechanism, the glycolytic enzyme phosphoglycerate kinase gets phosphorylated and, as a result, translocates into the mitochondrial matrix, where it acts as a protein kinase to phosphorylate PDK-1 and increases its catalytic activity. Thus, the enhanced activity of PDK-1 in cancer cells is the consequence of increased expression as well as post-translational modification. As a result, pyruvate is prevented from mitochondrial oxidation, thus getting diverted to lactate production in the cytoplasm (pyruvic acid + NADH + H
+ → lactic acid + NAD
+) (
Figure 1C). This drives aerobic glycolysis in cancer cells by providing NAD
+ for the reaction mediated by GAPDH.
Since aerobic glycolysis in cancer cells generates only 2 ATP per glucose instead of 32 ATP when glucose gets converted to CO2, it raises the question as to the energy status of the cancer cells. The unrestricted growth and proliferation of cancer cells cannot occur when the cells are energy-deficient. The energy status for supporting rapid growth is maintained in cancer cells by two mechanisms: one by accelerating “aerobic glycolysis” with an increased conversion of glucose to lactic acid and the other by generating energy from the metabolism of amino acids, primarily glutamine (Figure 1C). Even though glucose → lactic acid in cancer cells generates only two ATP per glucose, more ATP can be produced if the pathway is enhanced to metabolize more glucose. This metabolic switch together with glutamine-derived ATP maintains the energy status of the cancer cells to fuel their growth and proliferation.
Since glycolysis is subject to negative feedback regulation by ATP at the level of PFK-1, how can cancer cells enhance glycolysis and at the same time generate ATP at levels even higher than in normal cells? This is achieved by an increased production of fructose-2,6-bisphosphate, another regulatory molecule for PFK-1. While ATP is an inhibitor of PFK-1, fructose-2,6-bisphosphate is an activator that annuls the inhibition by ATP (
Figure 1C). The cellular levels of this activator are controlled by the bifunctional enzyme phosphofructokinase-2/fructose-2,6-bisphosphatase (PFKFB). Two isoforms of this enzyme, namely PFKFB3 and PFKFB4, are upregulated in cancer cells, leading to increased levels of fructose-2,6-bisphosphate to rescue glycolysis from the negative feedback regulation by ATP
[33][34]. The levels of fructose-2,6-bisphosphate in cancer cells are also regulated by another mechanism involving the protein TIGAR (TP53-induced glycolysis and apoptosis inhibitor)
[35]. The expression of TIGAR is downregulated in p53-mutant tumors. TIGAR possesses the catalytic activity of fructose-2,6-bisphosphatase and hence has the ability to degrade fructose-2,6-bisphosphate. This results in a reciprocal relationship between the levels of TIGAR and frunctose-2,6-bisphosphate. Since the expression of TIGAR is decreased in p53-deficient tumors, fructose-2,6-bisphosphate levels go up to maintain “aerobic glycolysis” by keeping PFK-1 active even in the presence of ATP.
There are two structurally distinct genes coding for LDH: LDH-A and LDH-B. Since the holoenzyme is a tetramer, LDH exists in five different isoforms depending on the composition of the two gene products in the tetramer. LDH1 consists of all four subunits being LDH-B whereas LDH5 consists of all four subunits being LDH-A. LDH2, LDH3, and LDH4 consist of varying mixtures of both LDH-A and LDH-B. The relative affinities of LDH-A and LDH-B for lactate and pyruvate make LDH-A more amenable to facilitate the conversion of pyruvate to lactate and LDH-B more amenable to facilitate the conversion of lactate to pyruvate
[36]. The expression of LDH-A is upregulated in all cancers whereas the expression of LDH-B is downregulated in most cancers. This shift in the relative amounts of the two isoforms facilitates “aerobic glycolysis” in cancer cells to convert pyruvate to lactate.
3.2. Aerobic Glycolysis in Non-Malignant Cells and Anerobic Glycolysis in Malignant Cells
Interestingly, aerobic glycolysis, where the conversion of glucose to lactic acid occurs even in the presence of adequate oxygen, is observed in certain cell types unrelated to cancer. This phenomenon may or may not be connected to a high cell proliferation rate. The first example in this category is the role of astrocytes in the brain and retina as providers of metabolic fuel to neurons in the form of lactate
[37]. Glucose is metabolized in astrocytes primarily to produce lactic acid, which is then supplied to neurons as an energy substrate; this process is not related to an increased proliferation of astrocytes. Another example is immune cells (monocytes and lymphocytes) during the adaptive response
[38]. In this case, the purpose of aerobic glycolysis may be to generate ATP at a rapid rate and also regulate certain specific signaling metabolites (e.g., lactate, succinate). This happens in activated lymphocytes, including those present in the context of a tumor to mount an immune response against the tumor cells, where aerobic glycolysis is associated with increased cell proliferation for the expansion of cytotoxic T cells. The same phenomenon also occurs in monocytes exposed to bacterial and fungal cell wall components. Another example is the endothelial cells involved in vessel sprouting, which opt for aerobic glycolysis with the generation of lactic acid
[38]. Here, lactate is likely to function as a signaling molecule to aid in various cellular processes necessary for the construction of new blood vessels. It is, however, interesting to note that the “stalk” cells and the “tip” cells associated with vessel sprouting do exhibit certain features, such as increased motility and invasion, similar to cancer cells.
Hypoxia and the resultant anaerobic glycolysis do occur in cancer cells present in solid tumors. The rate of proliferation of cancer cells exceeds the rate of formation of tumor-associated blood vessels. As a result, heterogeneity exists among cancer cells in terms of oxygen availability. Cancer cells that are located far away from blood vessels are subjected to hypoxia, and therefore these cells shift their glucose metabolism to anaerobic glycolysis as normal cells do under similar hypoxic conditions. But, metabolic reprogramming that occurs in cancer cells due to oncogenes and tumor suppressors may force these cells to continue to generate lactic acid from glucose even when an adequate blood supply becomes available to them, thus shifting from anaerobic glycolysis to aerobic glycolysis with the continued generation of lactic acid.
This entry is adapted from the peer-reviewed paper 10.3390/cancers16030504