DNA methylation is a fundamental mechanism of epigenetic control in cells and its dysregulation is strongly implicated in cancer development. Cancers possess an extensively hypomethylated genome with focal regions of hypermethylation at CPG islands. Due to the highly conserved nature of cancer-specific methylation, its detection in cell-free DNA in plasma using liquid biopsies constitutes an area of interest in biomarker research. The advent of next-generation sequencing and newer computational technologies have allowed for the development of diagnostic and prognostic biomarkers that utilize methylation profiling to diagnose disease and stratify risk. Methylome-based predictive biomarkers can determine the response to anti-cancer therapy. An additional emerging application of these biomarkers is in minimal residual disease monitoring. Several key challenges need to be addressed before cfDNA-based methylation biomarkers become fully integrated into practice. The first relates to the biology and stability of cfDNA. The second concerns the clinical validity and generalizability of methylation-based assays, many of which are cancer type-specific. The third involves their practicability, which is a stumbling block for translating technologies from bench to clinic.
- DNA methylation
- liquid biopsy
- biomarkers
- cell-free DNA
- 5-methylcytosine
1. Introduction
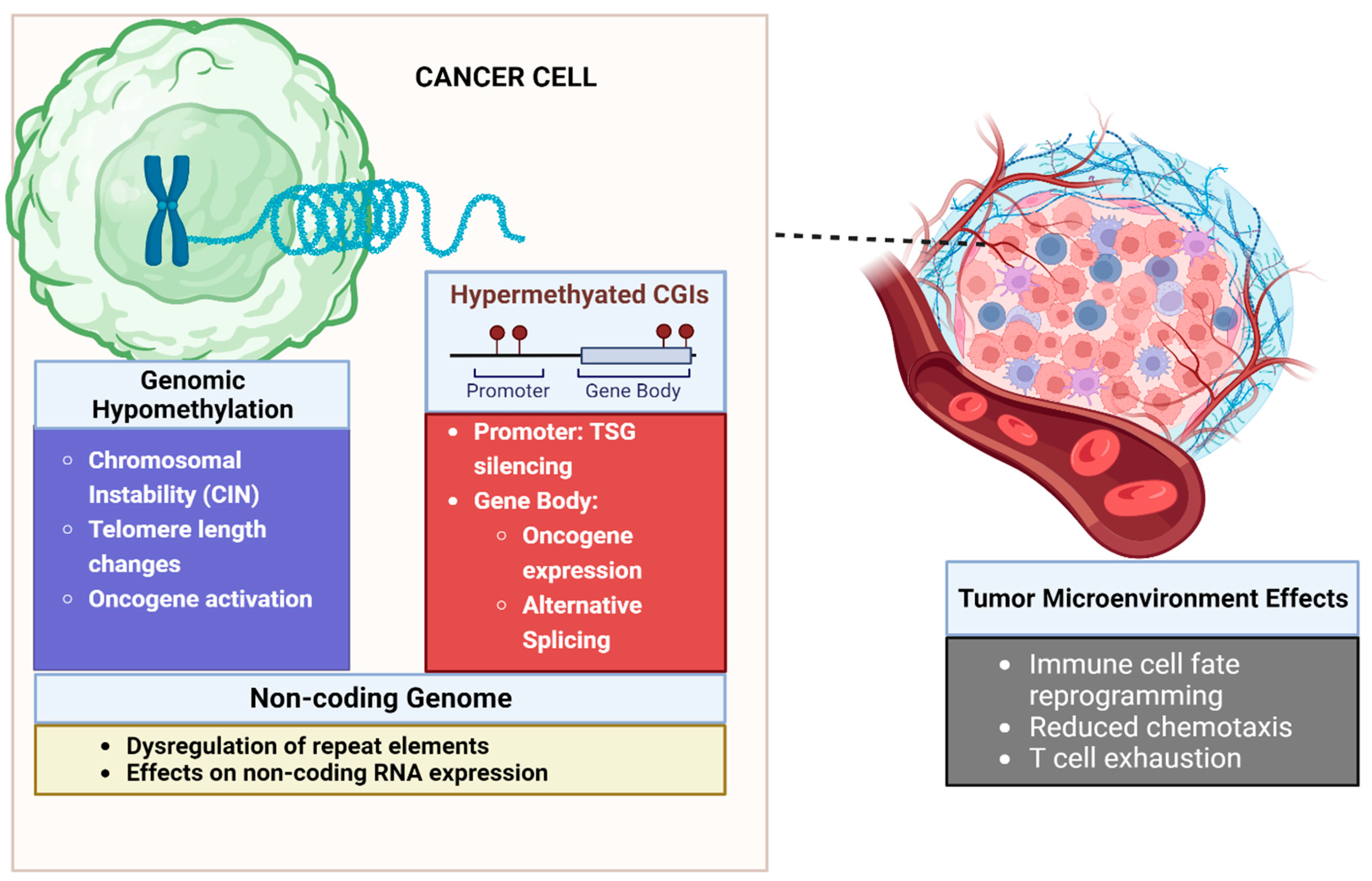
2. Biology of DNA Methylation in Malignancy
2.1. Aberrant DNA Methylation of CpG Islands Are Seen in Cancer
2.2. Gene Promoter Hypermethylation Leads to Silencing of Tumor Suppressor Genes
2.3. Methylation Can Lead to Epigenetic Activation of Oncogenic Pathways
2.4. Global DNA Hypomethylation Leads to Chromosomal Instability
2.5. DNA Methylation Influences Gene Expression beyond Promoter Silencing
2.6. Methylation Can Influence the Non-Coding Genome, Leading to Cancer
2.7. Role of DNA Methylation in the Tumor Microenvironment
3. Cell-Free Tumor Methylome Recapitulates the Cancer Epigenome
4. Applications of cfDNA Methylation as Biomarker in Cancer
4.1. Methylation as a Diagnostic Biomarker
4.2. Methylation as a Prognostic Biomarker of Patient Outcomes
4.3. Methylation as a Predictive Biomarker of Treatment Response
4.4. Minimal Residual Disease
This entry is adapted from the peer-reviewed paper 10.3390/curroncol31010033
References
- Wang, Q.; Xiong, F.; Wu, G.; Liu, W.; Chen, J.; Wang, B.; Chen, Y. Gene Body Methylation in Cancer: Molecular Mechanisms and Clinical Applications. Clin. Epigenetics 2022, 14, 154.
- Nishiyama, A.; Nakanishi, M. Navigating the DNA Methylation Landscape of Cancer. Trends Genet. 2021, 37, 1012–1027.
- Yokoyama, T.; Takehara, K.; Sugimoto, N.; Kaneko, K.; Fujimoto, E.; Okazawa-Sakai, M.; Okame, S.; Shiroyama, Y.; Yokoyama, T.; Teramoto, N.; et al. Lynch Syndrome-Associated Endometrial Carcinoma with MLH1 rmline Mutation and MLH1 Promoter Hypermethylation: A Case Report and Literature Review. BMC Cancer 2018, 18, 576.
- Joo, J.E.; Mahmood, K.; Walker, R.; Georgeson, P.; Candiloro, I.; Clendenning, M.; Como, J.; Joseland, S.; Preston, S.; Graversen, L.; et al. Identifying Primary and Secondary MLH1 Epimutation Carriers Displaying Low-Level Constitutional MLH1 Methylation Using Droplet Digital PCR and Genome-Wide DNA Methylation Profiling of Colorectal Cancers. Clin. Epigenetics 2023, 15, 95.
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505.
- Yates, J.; Boeva, V. Deciphering the Etiology and Role in Oncogenic Transformation of the CpG Island Methylator Phenotype: A Pan-Cancer Analysis. Brief. Bioinform. 2022, 23, bbab610.
- Romero-Garcia, S.; Prado-Garcia, H.; Carlos-Reyes, A. Role of DNA Methylation in the Resistance to Therapy in Solid Tumors. Front. Oncol. 2020, 10, 1152.
- McLachlan, T.; Matthews, W.C.; Jackson, E.R.; Staudt, D.E.; Douglas, A.M.; Findlay, I.J.; Persson, M.L.; Duchatel, R.J.; Mannan, A.; Germon, Z.P.; et al. B-Cell Lymphoma 6 (BCL6): From Master Regulator of Humoral Immunity to Oncogenic Driver in Pediatric Cancers. Mol. Cancer Res. 2022, 20, 1711–1723.
- Zhang, C.; Sheng, Q.; Zhao, N.; Huang, S.; Zhao, Y. DNA Hypomethylation Mediates Immune Response in Pan-Cancer. Epigenetics 2023, 18, 2192894.
- Ankill, J.; Aure, M.R.; Bjørklund, S.; Langberg, S.; Kristensen, V.N.; Vitelli, V.; Tekpli, X.; Fleischer, T. Epigenetic Alterations at Distal Enhancers Are Linked to Proliferation in Human Breast Cancer. NAR Cancer 2022, 4, zcac008.
- Cho, J.-W.; Shim, H.S.; Lee, C.Y.; Park, S.Y.; Hong, M.H.; Lee, I.; Kim, H.R. The Importance of Enhancer Methylation for Epigenetic Regulation of Tumorigenesis in Squamous Lung Cancer. Exp. Mol. Med. 2022, 54, 12–22.
- Pongor, L.S.; Tlemsani, C.; Elloumi, F.; Arakawa, Y.; Jo, U.; Gross, J.M.; Mosavarpour, S.; Varma, S.; Kollipara, R.K.; Roper, N.; et al. Integrative Epigenomic Analyses of Small Cell Lung Cancer Cells Demonstrates the Clinical Translational Relevance of Gene Body Methylation. iScience 2022, 25, 105338.
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene Body Methylation Can Alter Gene Expression and Is a Therapeutic Target in Cancer. Cancer Cell 2014, 26, 577–590.
- Feng, Y.; Zhang, T.; Wang, Y.; Xie, M.; Ji, X.; Luo, X.; Huang, W.; Xia, L. Homeobox Genes in Cancers: From Carcinogenesis to Recent Therapeutic Intervention. Front. Oncol. 2021, 11, 770428.
- Su, J.; Huang, Y.-H.; Cui, X.; Wang, X.; Zhang, X.; Lei, Y.; Xu, J.; Lin, X.; Chen, K.; Lv, J.; et al. Homeobox Oncogene Activation by Pan-Cancer DNA Hypermethylation. Genome Biol. 2018, 19, 108.
- Besselink, N.; Keijer, J.; Vermeulen, C.; Boymans, S.; de Ridder, J.; van Hoeck, A.; Cuppen, E.; Kuijk, E. The Genome-Wide Mutational Consequences of DNA Hypomethylation. Sci. Rep. 2023, 13, 6874.
- Chen, R.; Ishak, C.A.; De Carvalho, D.D. Endogenous Retroelements and the Viral Mimicry Response in Cancer Therapy and Cellular Homeostasis. Cancer Discov. 2021, 11, 2707–2725.
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618.
- Tire, B.; Ozturk, S. Potential Effects of Assisted Reproductive Technology on Telomere Length and Telomerase Activity in Human Oocytes and Early Embryos. J. Ovarian Res. 2023, 16, 130.
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46.
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; Caesar-Johnson, S.J.; Demchok, J.A.; Felau, I.; et al. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep. 2018, 23, 282–296.e4.
- Bonnal, S.C.; López-Oreja, I.; Valcárcel, J. Roles and Mechanisms of Alternative Splicing in Cancer—Implications for Care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474.
- Chen, Y.-C.; Elnitski, L. Aberrant DNA Methylation Defines Isoform Usage in Cancer, with Functional Implications. PLoS Comput. Biol. 2019, 15, e1007095.
- Jang, H.S.; Shah, N.M.; Du, A.Y.; Dailey, Z.Z.; Pehrsson, E.C.; Godoy, P.M.; Zhang, D.; Li, D.; Xing, X.; Kim, S.; et al. Transposable Elements Drive Widespread Expression of Oncogenes in Human Cancers. Nat. Genet. 2019, 51, 611–617.
- Espinet, E.; Gu, Z.; Imbusch, C.D.; Giese, N.A.; Büscher, M.; Safavi, M.; Weisenburger, S.; Klein, C.; Vogel, V.; Falcone, M.; et al. Aggressive PDACs Show Hypomethylation of Repetitive Elements and the Execution of an Intrinsic IFN Program Linked to a Ductal Cell of Origin. Cancer Discov. 2021, 11, 638–659.
- Tavora, B.; Mederer, T.; Wessel, K.J.; Ruffing, S.; Sadjadi, M.; Missmahl, M.; Ostendorf, B.N.; Liu, X.; Kim, J.-Y.; Olsen, O.; et al. Tumoural Activation of TLR3–SLIT2 Axis in Endothelium Drives Metastasis. Nature 2020, 586, 299–304.
- Liu, S.J.; Dang, H.X.; Lim, D.A.; Feng, F.Y.; Maher, C.A. Long Noncoding RNAs in Cancer Metastasis. Nat. Rev. Cancer 2021, 21, 446–460.
- Bunch, H. Gene Regulation of Mammalian Long Non-Coding RNA. Mol. Genet. Genom. 2018, 293, 1–15.
- Al-Imam, M.J.; Hussein, U.A.-R.; Sead, F.F.; Faqri, A.M.A.; Mekkey, S.M.; Khazel, A.J.; Almashhadani, H.A. The Interactions between DNA Methylation Machinery and Long Non-Coding RNAs in Tumor Progression and Drug Resistance. DNA Repair 2023, 128, 103526.
- Sideris, N.; Dama, P.; Bayraktar, S.; Stiff, T.; Castellano, L. LncRNAs in Breast Cancer: A Link to Future Approaches. Cancer Gene Ther. 2022, 29, 1866–1877.
- Shen, S.; Chen, J.; Li, H.; Jiang, Y.; Wei, Y.; Zhang, R.; Zhao, Y.; Chen, F. Large-Scale Integration of the Non-Coding RNAs with DNA Methylation in Human Cancers. Cell Rep. 2023, 42, 112261.
- Zhong, F.; Lin, Y.; Zhao, L.; Yang, C.; Ye, Y.; Shen, Z. Reshaping the Tumour Immune Microenvironment in Solid Tumours via Tumour Cell and Immune Cell DNA Methylation: From Mechanisms to Therapeutics. Br. J. Cancer 2023, 129, 24–37.
- Sun, L.; Su, Y.; Jiao, A.; Wang, X.; Zhang, B. T Cells in Health and Disease. Signal Transduct. Target. Ther. 2023, 8, 235.
- Henning, A.N.; Roychoudhuri, R.; Restifo, N.P. Epigenetic Control of CD8+ T Cell Differentiation. Nat. Rev. Immunol. 2018, 18, 340–356.
- Rodríguez-Ubreva, J.; Català-Moll, F.; Obermajer, N.; Álvarez-Errico, D.; Ramirez, R.N.; Company, C.; Vento-Tormo, R.; Moreno-Bueno, G.; Edwards, R.P.; Mortazavi, A.; et al. Prostaglandin E2 Leads to the Acquisition of DNMT3A-Dependent Tolerogenic Functions in Human Myeloid-Derived Suppressor Cells. Cell Rep. 2017, 21, 154–167.
- Liu, R.; Zhao, E.; Yu, H.; Yuan, C.; Abbas, M.N.; Cui, H. Methylation across the Central Dogma in Health and Diseases: New Therapeutic Strategies. Signal Transduct. Target. Ther. 2023, 8, 310.
- Wang, X.; Cao, Q.; Yu, L.; Shi, H.; Xue, B.; Shi, H. Epigenetic Regulation of Macrophage Polarization and Inflammation by DNA Methylation in Obesity. JCI Insight 2016, 1, e87748.
- Chen, C.; Liu, T.; Tang, Y.; Luo, G.; Liang, G.; He, W. Epigenetic Regulation of Macrophage Polarization in Wound Healing. Burns Trauma 2023, 11, tkac057.
- Vadevoo, S.M.P.; Gunassekaran, G.R.; Yoo, J.D.; Kwon, T.-H.; Hur, K.; Chae, S.; Lee, B. Epigenetic Therapy Reprograms M2-Type Tumor-Associated Macrophages into an M1-like Phenotype by Upregulating MiR-7083-5p. Front. Immunol. 2022, 13, 976196.
- Dangaj, D.; Bruand, M.; Grimm, A.J.; Ronet, C.; Barras, D.; Duttagupta, P.A.; Lanitis, E.; Duraiswamy, J.; Tanyi, J.L.; Benencia, F.; et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019, 35, 885–900.e10.
- Zheng, Y.; Wang, Z.; Wei, S.; Liu, Z.; Chen, G. Epigenetic Silencing of Chemokine CCL2 Represses Macrophage Infiltration to Potentiate Tumor Development in Small Cell Lung Cancer. Cancer Lett. 2021, 499, 148–163.
- Khan, P.; Fatima, M.; Khan, M.A.; Batra, S.K.; Nasser, M.W. Emerging Role of Chemokines in Small Cell Lung Cancer: Road Signs for Metastasis, Heterogeneity, and Immune Response. Semin. Cancer Biol. 2022, 87, 117–126.
- Chen, X.; Pan, X.; Zhang, W.; Guo, H.; Cheng, S.; He, Q.; Yang, B.; Ding, L. Epigenetic Strategies Synergize with PD-L1/PD-1 Targeted Cancer Immunotherapies to Enhance Antitumor Responses. Acta Pharm. Sin. B 2020, 10, 723–733.
- Prinzing, B.; Zebley, C.C.; Petersen, C.T.; Fan, Y.; Anido, A.A.; Yi, Z.; Nguyen, P.; Houke, H.; Bell, M.; Haydar, D.; et al. Deleting DNMT3A in CAR T Cells Prevents Exhaustion and Enhances Antitumor Activity. Sci. Transl. Med. 2023, 13, eabh0272.
- Yang, R.; Cheng, S.; Luo, N.; Gao, R.; Yu, K.; Kang, B.; Wang, L.; Zhang, Q.; Fang, Q.; Zhang, L.; et al. Distinct Epigenetic Features of Tumor-Reactive CD8+ T Cells in Colorectal Cancer Patients Revealed by Genome-Wide DNA Methylation Analysis. Genome Biol. 2019, 21, 2.
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid Biopsies Come of Age: Towards Implementation of Circulating Tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238.
- Gaitsch, H.; Franklin, R.J.M.; Reich, D.S. Cell-Free DNA-Based Liquid Biopsies in Neurology. Brain 2023, 146, 1758–1774.
- Corcoran, R.B.; Chabner, B.A. Application of Cell-Free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765.
- Lau, B.T.; Almeda, A.; Schauer, M.; McNamara, M.; Bai, X.; Meng, Q.; Partha, M.; Grimes, S.M.; Lee, H.; Heestand, G.M.; et al. Single-Molecule Methylation Profiles of Cell-Free DNA in Cancer with Nanopore Sequencing. Genome Med. 2023, 15, 33.
- Cescon, D.W.; Bratman, S.V.; Chan, S.M.; Siu, L.L. Circulating Tumor DNA and Liquid Biopsy in Oncology. Nat. Cancer 2020, 1, 276–290.
- Igari, F.; Tanaka, H.; Giuliano, A.E. The Applications of Plasma Cell-Free DNA in Cancer Detection: Implications in the Management of Breast Cancer Patients. Crit. Rev. Oncol. Hematol. 2022, 175, 103725.
- Smith, J.T.; Balar, A.; Lakhani, D.A.; Kluwe, C.; Zhao, Z.; Kopparapu, P.; Almodovar, K.; Muterspaugh, A.; Yan, Y.; York, S.; et al. Circulating Tumor DNA as a Biomarker of Radiographic Tumor Burden in SCLC. JTO Clin. Res. Rep. 2021, 2, 100110.
- Mohan, S.; Foy, V.; Ayub, M.; Leong, H.S.; Schofield, P.; Sahoo, S.; Descamps, T.; Kilerci, B.; Smith, N.K.; Carter, M.; et al. Profiling of Circulating Free DNA Using Targeted and Genome-Wide Sequencing in Patients with SCLC. J. Thorac. Oncol. 2020, 15, 216–230.
- Almodovar, K.; Iams, W.T.; Meador, C.B.; Zhao, Z.; York, S.; Horn, L.; Yan, Y.; Hernandez, J.; Chen, H.; Shyr, Y.; et al. Longitudinal Cell-Free DNA Analysis in Patients with Small Cell Lung Cancer Reveals Dynamic Insights into Treatment Efficacy and Disease Relapse. J. Thorac. Oncol. 2018, 13, 112–123.
- Tolmeijer, S.H.; Boerrigter, E.; Sumiyoshi, T.; Kwan, E.M.; Ng, S.W.S.; Annala, M.; Donnellan, G.; Herberts, C.; Benoist, G.E.; Hamberg, P.; et al. Early On-Treatment Changes in Circulating Tumor DNA Fraction and Response to Enzalutamide or Abiraterone in Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2023, 29, 2835–2844.
- Semenkovich, N.P.; Szymanski, J.J.; Earland, N.; Chauhan, P.S.; Pellini, B.; Chaudhuri, A.A. Genomic Approaches to Cancer and Minimal Residual Disease Detection Using Circulating Tumor DNA. J. Immunother. Cancer 2023, 11, e006284.
- Gatto, L.; Franceschi, E.; Di Nunno, V.; Tosoni, A.; Lodi, R.; Brandes, A.A. Liquid Biopsy in Glioblastoma Management: From Current Research to Future Perspectives. Oncologist 2021, 26, 865–878.
- Mair, R.; Mouliere, F. Cell-Free DNA Technologies for the Analysis of Brain Cancer. Br. J. Cancer 2022, 126, 371–378.
- Liu, L.; Toung, J.M.; Jassowicz, A.F.; Vijayaraghavan, R.; Kang, H.; Zhang, R.; Kruglyak, K.M.; Huang, H.J.; Hinoue, T.; Shen, H.; et al. Targeted Methylation Sequencing of Plasma Cell-Free DNA for Cancer Detection and Classification. Ann. Oncol. 2018, 29, 1445–1453.
- Sadeh, R.; Sharkia, I.; Fialkoff, G.; Rahat, A.; Gutin, J.; Chappleboim, A.; Nitzan, M.; Fox-Fisher, I.; Neiman, D.; Meler, G.; et al. ChIP-Seq of Plasma Cell-Free Nucleosomes Identifies Gene Expression Programs of the Cells of Origin. Nat. Biotechnol. 2021, 39, 586–598.
- Baca, S.C.; Seo, J.-H.; Davidsohn, M.P.; Fortunato, B.; Semaan, K.; Sotudian, S.; Lakshminarayanan, G.; Diossy, M.; Qiu, X.; El Zarif, T.; et al. Liquid Biopsy Epigenomic Profiling for Cancer Subtyping. Nat. Med. 2023, 29, 2737–2741.
- Nassiri, F.; Chakravarthy, A.; Feng, S.; Shen, S.Y.; Nejad, R.; Zuccato, J.A.; Voisin, M.R.; Patil, V.; Horbinski, C.; Aldape, K.; et al. Detection and Discrimination of Intracranial Tumors Using Plasma Cell-Free DNA Methylomes. Nat. Med. 2020, 26, 1044–1047.
- Burgener, J.M.; Zou, J.; Zhao, Z.; Zheng, Y.; Shen, S.Y.; Huang, S.H.; Keshavarzi, S.; Xu, W.; Liu, F.-F.; Liu, G.; et al. Tumor-Naïve Multimodal Profiling of Circulating Tumor DNA in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2021, 27, 4230–4244.
- Stackpole, M.L.; Zeng, W.; Li, S.; Liu, C.-C.; Zhou, Y.; He, S.; Yeh, A.; Wang, Z.; Sun, F.; Li, Q.; et al. Cost-Effective Methylome Sequencing of Cell-Free DNA for Accurately Detecting and Locating Cancer. Nat. Commun. 2022, 13, 5566.
- Galardi, F.; De Luca, F.; Romagnoli, D.; Biagioni, C.; Moretti, E.; Biganzoli, L.; Di Leo, A.; Migliaccio, I.; Malorni, L.; Benelli, M. Cell-Free DNA-Methylation-Based Methods and Applications in Oncology. Biomolecules 2020, 10, 1677.
- Fu, S.; Debes, J.D.; Boonstra, A. DNA Methylation Markers in the Detection of Hepatocellular Carcinoma. Eur. J. Cancer 2023, 191, 112960.
- Luo, H.; Wei, W.; Ye, Z.; Zheng, J.; Xu, R. Liquid Biopsy of Methylation Biomarkers in Cell-Free DNA. Trends Mol. Med. 2021, 27, 482–500.
- Shen, S.Y.; Burgener, J.M.; Bratman, S.V.; De Carvalho, D.D. Preparation of CfMeDIP-Seq Libraries for Methylome Profiling of Plasma Cell-Free DNA. Nat. Protoc. 2019, 14, 2749–2780.
- deVos, T.; Tetzner, R.; Model, F.; Weiss, G.; Schuster, M.; Distler, J.; Steiger, K.V.; Grutzmann, R.; Pilarsky, C.; Habermann, J.K.; et al. Circulating Methylated SEPT9 DNA in Plasma Is a Biomarker for Colorectal Cancer. Clin. Chem. 2009, 55, 1337–1346.
- Müller, D.; Győrffy, B. DNA Methylation-Based Diagnostic, Prognostic, and Predictive Biomarkers in Colorectal Cancer. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2022, 1877, 188722.
- Weiss, G.; Schlegel, A.; Kottwitz, D.; König, T.; Tetzner, R. Validation of the SHOX2/PTGER4 DNA Methylation Marker Panel for Plasma-Based Discrimination between Patients with Malignant and Nonmalignant Lung Disease. J. Thorac. Oncol. 2017, 12, 77–84.
- Oh, T.; Kim, N.; Moon, Y.; Kim, M.S.; Hoehn, B.D.; Park, C.H.; Kim, T.S.; Kim, N.K.; Chung, H.C.; An, S. Genome-Wide Identification and Validation of a Novel Methylation Biomarker, SDC2, for Blood-Based Detection of Colorectal Cancer. J. Mol. Diagn. 2013, 15, 498–507.
- Klein, E.A.; Madhavan, S.; Beer, T.M.; Bettegowda, C.; Liu, M.C.; Hartman, A.-R.; Hackshaw, A. Dying to Find Out: The Cost of Time at the Dawn of the Multicancer Early Detection Era. Cancer Epidemiol. Biomark. Prev. 2023, 32, 1003–1010.
- Park, B.H.; Shen, S.Y.; Min, J.; Fleshner, N.; Knox, J.; May, T.; Ailles, L.; Newton, Y.; Zhang, J.; Singhania, R. Development of a Genome-Wide Methylome Enrichment Platform for Multi-Cancer Early Detection (MCED). Cancer Res. 2023, 83 (Suppl. S7), 1030.
- Huang, M.; He, J.; Lai, W.; Liu, L.; Xu, H.; Zeng, Y.; Lan, Q.; Lin, X.; Chu, Z. Methylated Septin 9 Gene Is an Important Prognostic Marker in Stage II and Stage III Colorectal Cancer for Evaluating Local Recurrence or Distant Metastasis after Surgery. BMC Gastroenterol. 2022, 22, 87.
- Hier, J.; Vachon, O.; Bernstein, A.; Ibrahim, I.; Mlynarek, A.; Hier, M.; Alaoui-Jamali, M.A.; Maschietto, M.; da Silva, S.D. Portrait of DNA Methylated Genes Predictive of Poor Prognosis in Head and Neck Cancer and the Implication for Targeted Therapy. Sci. Rep. 2021, 11, 10012.
- Ko, K.; Kananazawa, Y.; Yamada, T.; Kakinuma, D.; Matsuno, K.; Ando, F.; Kuriyama, S.; Matsuda, A.; Yoshida, H. Methylation Status and Long-Fragment Cell-Free DNA Are Prognostic Biomarkers for Gastric Cancer. Cancer Med. 2021, 10, 2003–2012.
- Bae, J.M.; Shin, S.-H.; Kwon, H.-J.; Park, S.-Y.; Kook, M.C.; Kim, Y.-W.; Cho, N.-Y.; Kim, N.; Kim, T.-Y.; Kim, D.; et al. ALU and LINE-1 Hypomethylations in Multistep Gastric Carcinogenesis and Their Prognostic Implications. Int. J. Cancer 2012, 131, 1323–1331.
- Ul Haq, S.; Schmid, S.; Aparnathi, M.K.; Hueniken, K.; Zhan, L.J.; Sacdalan, D.; Li, J.J.N.; Meti, N.; Patel, D.; Cheng, D.; et al. Cell-Free DNA Methylation-Defined Prognostic Subgroups in Small-Cell Lung Cancer Identified by Leukocyte Methylation Subtraction. iScience 2022, 25, 105487.
- Rini, B.I.; Zhang, J.; Hall, O.; Bergener, J.; Wang, Y.; Brown, B.; Min, J.; Shen, S.Y.; Fleshner, N.; Polio, A.; et al. 1910P Evaluation of a Genome-Wide Methylome Enrichment Platform for Circulating Tumor DNA Quantification and Prognostic Performance in Renal Cell Carcinoma (RCC). Ann. Oncol. 2023, 34, S1028.
- Liu, G.; Zhang, J.; Hall, O.; Bergener, J.; Wang, Y.; Brown, B.; Min, J.; Shen, S.Y.; Pienkowski, M.; Huang, S.H.; et al. 866P Prognostic Performance of a Genome-Wide Methylome Enrichment Platform in Head and Neck Cancer. Ann. Oncol. 2023, 34, S561.
- Majchrzak-Celińska, A.; Paluszczak, J.; Kleszcz, R.; Magiera, M.; Barciszewska, A.-M.; Nowak, S.; Baer-Dubowska, W. Detection of MGMT, RASSF1A, P15INK4B, and P14ARF Promoter Methylation in Circulating Tumor-Derived DNA of Central Nervous System Cancer Patients. J. Appl. Genet. 2013, 54, 335–344.
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; de Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N.; et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 523–535.
- He, T.; Zhang, M.; Zheng, R.; Zheng, S.; Linghu, E.; Herman, J.G.; Guo, M. Methylation of SLFN11 Is a Marker of Poor Prognosis and Cisplatin Resistance in Colorectal Cancer. Epigenomics 2017, 9, 849–862.
- Stewart, C.A.; Tong, P.; Cardnell, R.J.; Sen, T.; Li, L.; Gay, C.M.; Masrorpour, F.; Fan, Y.; Bara, R.O.; Feng, Y. Dynamic Variations in Epithelial-to-Mesenchymal Transition (EMT), ATM, and SLFN11 Govern Response to PARP Inhibitors and Cisplatin in Small Cell Lung Cancer. Oncotarget 2017, 8, 28575.
- Murai, J.; Feng, Y.; Guoying, K.Y.; Ru, Y.; Tang, S.-W.; Shen, Y.; Pommier, Y. Resistance to PARP Inhibitors by SLFN11 Inactivation Can Be Overcome by ATR Inhibition. Oncotarget 2016, 7, 76534.
- Tserpeli, V.; Stergiopoulou, D.; Londra, D.; Giannopoulou, L.; Buderath, P.; Balgkouranidou, I.; Xenidis, N.; Grech, C.; Obermayr, E.; Zeillinger, R. Prognostic Significance of SLFN11 Methylation in Plasma Cell-Free DNA in Advanced High-Grade Serous Ovarian Cancer. Cancers 2021, 14, 4.
- André, T.; Cohen, R.; Salem, M.E. Immune Checkpoint Blockade Therapy in Patients with Colorectal Cancer Harboring Microsatellite Instability/Mismatch Repair Deficiency in 2022. In American Society of Clinical Oncology Educational Book; American Society of Clinical Oncology: Alexandria, VA, USA, 2022; Volume 42, pp. 233–241.
- Pasanen, A.; Loukovaara, M.; Bützow, R. Clinicopathological Significance of Deficient DNA Mismatch Repair and MLH1 Promoter Methylation in Endometrioid Endometrial Carcinoma. Mod. Pathol. 2020, 33, 1443–1452.
- Wang, D.; O’Rourke, D.; Sanchez-Garcia, J.F.; Cai, T.; Scheuenpflug, J.; Feng, Z. Development of a Liquid Biopsy Based Purely Quantitative Digital Droplet PCR Assay for Detection of MLH1 Promoter Methylation in Colorectal Cancer Patients. BMC Cancer 2021, 21, 797.
- Lu, Y.-T.; Plets, M.; Morrison, G.; Cunha, A.T.; Cen, S.Y.; Rhie, S.K.; Siegmund, K.D.; Daneshmand, S.; Quinn, D.I.; Meeks, J.J.; et al. Cell-Free DNA Methylation as a Predictive Biomarker of Response to Neoadjuvant Chemotherapy for Patients with Muscle-Invasive Bladder Cancer in SWOG S1314. Eur. Urol. Oncol. 2023, 6, 516–524.
- Luskin, M.R.; Murakami, M.A.; Manalis, S.R.; Weinstock, D.M. Targeting Minimal Residual Disease: A Path to Cure? Nat. Rev. Cancer 2018, 18, 255–263.
- Norton, L. Cancer Log-Kill Revisited. In American Society of Clinical Oncology Educational Book; American Society of Clinical Oncology: Alexandria, VA, USA, 2014; Volume 34, pp. 3–7.
- Blackburn, E.H. Cancer Interception. Cancer Prev. Res. 2011, 4, 787–792.
- Chen, X.; Zhang, J.; Ruan, W.; Huang, M.; Wang, C.; Wang, H.; Jiang, Z.; Wang, S.; Liu, Z.; Liu, C.; et al. Urine DNA Methylation Assay Enables Early Detection and Recurrence Monitoring for Bladder Cancer. J. Clin. Investig. 2020, 130, 6278–6289.
- van Zogchel, L.M.J.; Lak, N.S.M.; Verhagen, O.J.H.M.; Tissoudali, A.; Gussmalla Nuru, M.; Gelineau, N.U.; Zappeij-Kannengieter, L.; Javadi, A.; Zijtregtop, E.A.M.; Merks, J.H.M.; et al. Novel Circulating Hypermethylated RASSF1A DdPCR for Liquid Biopsies in Patients with Pediatric Solid Tumors. JCO Precis. Oncol. 2021, 5, 1738–1748.
- Yuan, Z.; Wang, S.; Ni, K.; Zhan, Y.; Ma, H.; Liu, X.; Xin, R.; Zhou, X.; Liu, Z.; Zhao, X. Circulating Methylated SEPT9 DNA Analyses to Predict Recurrence Risk and Adjuvant Chemotherapy Benefit in Stage II to III Colorectal Cancer. Med. Sci. Monit. 2022, 28, e937757-1.
- Mo, S.; Ye, L.; Wang, D.; Han, L.; Zhou, S.; Wang, H.; Dai, W.; Wang, Y.; Luo, W.; Wang, R.; et al. Early Detection of Molecular Residual Disease and Risk Stratification for Stage I to III Colorectal Cancer via Circulating Tumor DNA Methylation. JAMA Oncol. 2023, 9, 770–778.
- Leon Arellano, M.; García-Arranz, M.; Guadalajara, H.; Olivera-Salazar, R.; Valdes-Sanchez, T.; García-Olmo, D. Analysis of Septin 9 Gene Hypermethylation as Follow-Up Biomarker of Colorectal Cancer Patients after Curative Surgery. Diagnostics 2022, 12, 993.