Diabetes mellitus, the most prevalent endocrine disorder, not only impacts the retina but also significantly involves the ocular surface. Diabetes contributes to the development of dry eye disease and induces morphological and functional corneal alterations, particularly affecting nerves and epithelial cells. These changes manifest as epithelial defects, reduced sensitivity, and delayed wound healing, collectively encapsulated in the context of diabetic keratopathy. In advanced stages of this condition, the progression to corneal ulcers and scarring further unfolds, eventually leading to corneal opacities. This critical complication hampers vision and carries the potential for irreversible visual loss.
- diabetic complication
- cornea
- keratopathy
- redox
- pathways
- molecular
- targets
1. Introduction
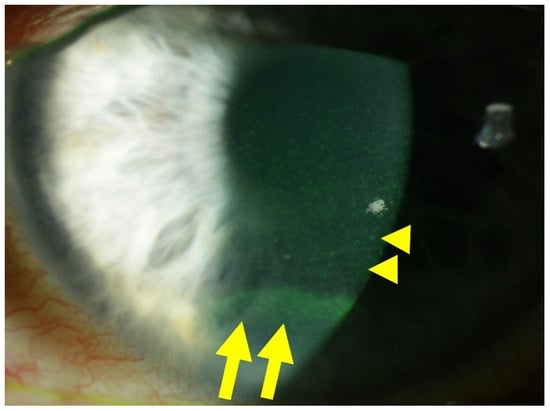
2. Corneal Cell Damage in Diabetic Keratopathy
2.1. Redox Signaling in the Diabetic Cornea
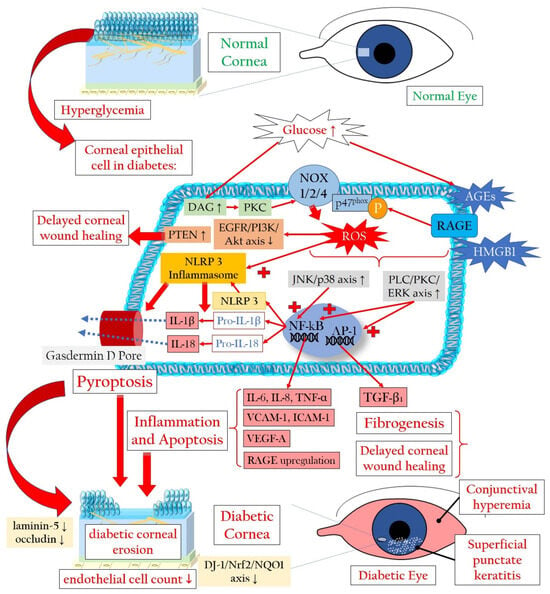
2.2. Pyroptosis in the Corneal Epithelium Triggered by Hyperglycemia
2.3. Involvement of Stroma, Endothelium, and Tight Junctions in Diabetic Damage
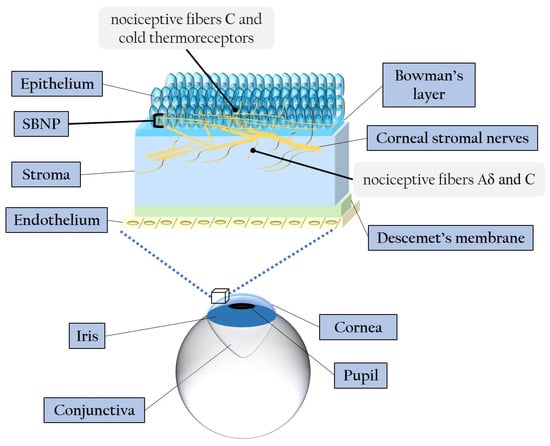
3. Diabetic Corneal Neuropathy
3.1. Evidence of Corneal Nerve Damage in Diabetes
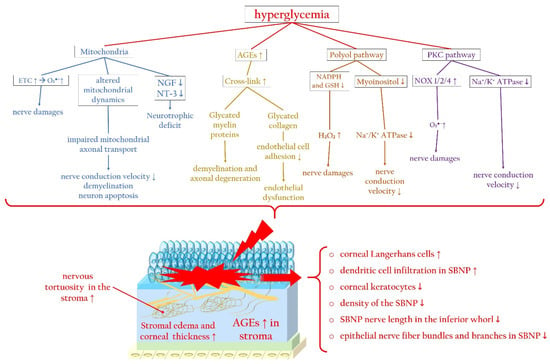
3.2. Molecular Pathways Leading to Corneal Nerve Damage
Hyperglycemia-Related Mitochondrial Dysfunction
AGEs and Axonal Degeneration
Effects of the Polyol Pathway and PKC Cascade
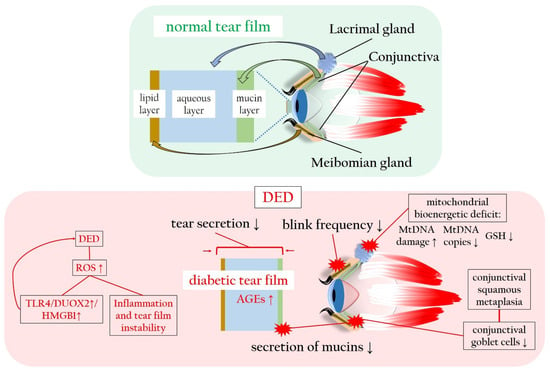
4. Diabetes-Associated Dry Eye Disease Promotes Oxidatives Stress and Inflammation
This entry is adapted from the peer-reviewed paper 10.3390/antiox13010120
References
- Ljubimov, A.V. Diabetic complications in the cornea. Vis. Res. 2017, 139, 138–152.
- Markoulli, M.; Flanagan, J.; Tummanapalli, S.S.; Wu, J.; Willcox, M. The impact of diabetes on corneal nerve morphology and ocular surface integrity. Ocul. Surf. 2018, 16, 45–57.
- Murphy, P.J.; Patel, S.; Kong, N.; Ryder, R.E.; Marshall, J. Noninvasive assessment of corneal sensitivity in young and elderly diabetic and nondiabetic subjects. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1737–1742.
- Mukhija, R.; Gupta, N.; Vashist, P.; Tandon, R.; Gupta, S.K. Population-based assessment of visual impairment and pattern of corneal disease: Results from the CORE (Corneal Opacity Rural Epidemiological) study. Br. J. Ophthalmol. 2020, 104, 994–998.
- Chang, Y.-S.; Tai, M.-C.; Ho, C.-H.; Chu, C.-C.; Wang, J.-J.; Tseng, S.-H.; Jan, R.-L. Risk of Corneal Ulcer in Patients with Diabetes Mellitus: A Retrospective Large-Scale Cohort Study. Sci. Rep. 2020, 10, 7388.
- Friend, J.; Snip, R.C.; Kiorpes, T.C.; Thoft, R.A. Insulin sensitivity and sorbitol production of the normal rabbit corneal epithelium in vitro. Investig. Ophthalmol. Vis. Sci. 1980, 19, 913–919.
- Cunha, D.A.; Carneiro, E.M.; Alves, M.d.C.; Jorge, A.G.; Sousa, S.M.d.; Boschero, A.C.; Saad, M.J.A.; Velloso, L.A.; Rocha, E.M. Insulin secretion by rat lachrymal glands: Effects of systemic and local variables. Am. J. Physiol.-Endocrinol. Metab. 2005, 289, E768–E775.
- Mueckler, M. Family of glucose-transporter genes. Implications for glucose homeostasis and diabetes. Diabetes 1990, 39, 6–11.
- Stuard, W.L.; Titone, R.; Robertson, D.M. The IGF/Insulin-IGFBP Axis in Corneal Development, Wound Healing, and Disease. Front. Endocrinol. 2020, 11, 24.
- Zhu, L.; Titone, R.; Robertson, D.M. The impact of hyperglycemia on the corneal epithelium: Molecular mechanisms and insight. Ocul. Surf. 2019, 17, 644–654.
- Babizhayev, M.A.; Strokov, I.A.; Nosikov, V.V.; Savel’yeva, E.L.; Sitnikov, V.F.; Yegorov, Y.E.; Lankin, V.Z. The Role of Oxidative Stress in Diabetic Neuropathy: Generation of Free Radical Species in the Glycation Reaction and Gene Polymorphisms Encoding Antioxidant Enzymes to Genetic Susceptibility to Diabetic Neuropathy in Population of Type I Diabetic Patients. Cell Biochem. Biophys. 2015, 71, 1425–1443.
- González, P.; Lozano, P.; Ros, G.; Solano, F. Hyperglycemia and Oxidative Stress: An Integral, Updated and Critical Overview of Their Metabolic Interconnections. Int. J. Mol. Sci. 2023, 24, 9352.
- Ramya, R.; Coral, K.; Bharathidevi, S.R. RAGE silencing deters CML-AGE induced inflammation and TLR4 expression in endothelial cells. Exp. Eye Res. 2021, 206, 108519.
- Buonfiglio, F.; Böhm, E.W.; Pfeiffer, N.; Gericke, A. Oxidative Stress: A Suitable Therapeutic Target for Optic Nerve Diseases? Antioxidants 2023, 12, 1465.
- Böhm, E.W.; Buonfiglio, F.; Voigt, A.M.; Bachmann, P.; Safi, T.; Pfeiffer, N.; Gericke, A. Oxidative stress in the eye and its role in the pathophysiology of ocular diseases. Redox Biol. 2023, 68, 102967.
- Kim, J.; Kim, C.-S.; Sohn, E.; Jeong, I.-H.; Kim, H.; Kim, J.S. Involvement of advanced glycation end products, oxidative stress and nuclear factor-kappaB in the development of diabetic keratopathy. Graefe’s Arch. Clin. Exp. Ophthalmol. 2011, 249, 529–536.
- Shi, L.; Yu, X.; Yang, H.; Wu, X. Advanced Glycation End Products Induce Human Corneal Epithelial Cells Apoptosis through Generation of Reactive Oxygen Species and Activation of JNK and p38 MAPK Pathways. PLoS ONE 2013, 8, e66781.
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142.
- Chen, Y.-H.; Chen, Z.-W.; Li, H.-M.; Yan, X.-F.; Feng, B. AGE/RAGE-induced EMP release via the NOX-derived ROS pathway. J. Diabetes Res. 2018, 2018, 6823058.
- Dobi, A.; Bravo, S.B.; Veeren, B.; Paradela-Dobarro, B.; Álvarez, E.; Meilhac, O.; Viranaicken, W.; Baret, P.; Devin, A.; Rondeau, P. Advanced glycation end-products disrupt human endothelial cells redox homeostasis: New insights into reactive oxygen species production. Free Radic. Res. 2019, 53, 150–169.
- Hink, U.; Li, H.; Mollnau, H.; Oelze, M.; Matheis, E.; Hartmann, M.; Skatchkov, M.; Thaiss, F.; Stahl, R.A.K.; Warnholtz, A.; et al. Mechanisms Underlying Endothelial Dysfunction in Diabetes Mellitus. Circ. Res. 2001, 88, e14–e22.
- Giordo, R.; Nasrallah, G.K.; Posadino, A.M.; Galimi, F.; Capobianco, G.; Eid, A.H.; Pintus, G. Resveratrol-Elicited PKC Inhibition Counteracts NOX-Mediated Endothelial to Mesenchymal Transition in Human Retinal Endothelial Cells Exposed to High Glucose. Antioxidants 2021, 10, 224.
- Kolczynska, K.; Loza-Valdes, A.; Hawro, I.; Sumara, G. Diacylglycerol-evoked activation of PKC and PKD isoforms in regulation of glucose and lipid metabolism: A review. Lipids Health Dis. 2020, 19, 113.
- Yu, W.; Tao, M.; Zhao, Y.; Hu, X.; Wang, M. 4′-Methoxyresveratrol Alleviated AGE-Induced Inflammation via RAGE-Mediated NF-κB and NLRP3 Inflammasome Pathway. Molecules 2018, 23, 1447.
- Xu, K.P.; Li, Y.; Ljubimov, A.V.; Yu, F.S.X. High glucose suppresses epidermal growth factor receptor/phosphatidylinositol 3-kinase/Akt signaling pathway and attenuates corneal epithelial wound healing. Diabetes 2009, 58, 1077–1085.
- Taguchi, K.; Fukami, K. RAGE signaling regulates the progression of diabetic complications. Front. Pharmacol. 2023, 14, 1128872.
- Carrington, L.M.; Albon, J.; Anderson, I.; Kamma, C.; Boulton, M. Differential Regulation of Key Stages in Early Corneal Wound Healing by TGF-β Isoforms and Their Inhibitors. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1886–1894.
- Hou, Y.; Lan, J.; Zhang, F.; Wu, X. Expression profiles and potential corneal epithelial wound healing regulation targets of high-mobility group box 1 in diabetic mice. Exp. Eye Res. 2021, 202, 108364.
- Wan, L.; Bai, X.; Zhou, Q.; Chen, C.; Wang, H.; Liu, T.; Xue, J.; Wei, C.; Xie, L. The advanced glycation end-products (AGEs)/ROS/NLRP3 inflammasome axis contributes to delayed diabetic corneal wound healing and nerve regeneration. Int. J. Biol. Sci. 2022, 18, 809–825.
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128.
- Wooff, Y.; Fernando, N.; Wong, J.H.C.; Dietrich, C.; Aggio-Bruce, R.; Chu-Tan, J.A.; Robertson, A.A.B.; Doyle, S.L.; Man, S.M.; Natoli, R. Caspase-1-dependent inflammasomes mediate photoreceptor cell death in photo-oxidative damage-induced retinal degeneration. Sci. Rep. 2020, 10, 2263.
- Evavold, C.L.; Hafner-Bratkovič, I.; Devant, P.; D’Andrea, J.M.; Ngwa, E.M.; Boršić, E.; Doench, J.G.; LaFleur, M.W.; Sharpe, A.H.; Thiagarajah, J.R. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell 2021, 184, 4495–4511.e4419.
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307.
- Mauro, A.G.; Hunter, K.; Salloum, F.N. Chapter Five—Cardiac complications of cancer therapies. In Advances in Cancer Research; Gewirtz, D.A., Fisher, P.B., Eds.; Academic Press: Cambridge, MA, USA, 2022; Volume 155, pp. 167–214.
- Bitirgen, G.; Ozkagnici, A.; Malik, R.; Kerimoglu, H. Corneal nerve fibre damage precedes diabetic retinopathy in patients with type 2 diabetes mellitus. Diabet. Med. 2014, 31, 431–438.
- Zhang, X.; Qiu, J.; Huang, F.; Shan, K.; Zhang, C. Type 2 Diabetes Mellitus Makes Corneal Endothelial Cells Vulnerable to Ultraviolet A-Induced Oxidative Damage Via Decreased DJ-1/Nrf2/NQO1 Pathway. Invest. Ophthalmol. Vis. Sci. 2022, 63, 25.
- Xu, J.J.; Cui, J.; Lin, Q.; Chen, X.Y.; Zhang, J.; Gao, E.H.; Wei, B.; Zhao, W. Protection of the enhanced Nrf2 deacetylation and its downstream transcriptional activity by SIRT1 in myocardial ischemia/reperfusion injury. Int. J. Cardiol. 2021, 342, 82–93.
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal 2018, 28, 643–661.
- Wang, M.X.; Zhao, J.; Zhang, H.; Li, K.; Niu, L.Z.; Wang, Y.P.; Zheng, Y.J. Potential Protective and Therapeutic Roles of the Nrf2 Pathway in Ocular Diseases: An Update. Oxid. Med. Cell Longev. 2020, 2020, 9410952.
- Lu, W.; Ebihara, N.; Miyazaki, K.; Murakami, A. Reduced Expression of Laminin-5 in Corneal Epithelial Cells Under High Glucose Condition. Cornea 2006, 25, 61–67.
- Huang, C.; Liao, R.; Wang, F.; Tang, S. Characteristics of Reconstituted Tight Junctions After Corneal Epithelial Wounds and Ultrastructure Alterations of Corneas in Type 2 Diabetic Rats. Curr. Eye Res. 2016, 41, 783–790.
- Wang, Y.; Zhao, X.; Shi, D.; Chen, P.; Yu, Y.; Yang, L.; Xie, L. Overexpression of SIRT1 Promotes High Glucose–Attenuated Corneal Epithelial Wound Healing via p53 Regulation of the IGFBP3/IGF-1R/AKT Pathway. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3806–3814.
- Jiang, Q.-W.; Kaili, D.; Freeman, J.; Lei, C.-Y.; Geng, B.-C.; Tan, T.; He, J.-F.; Shi, Z.; Ma, J.-J.; Luo, Y.-H. Diabetes inhibits corneal epithelial cell migration and tight junction formation in mice and human via increasing ROS and impairing Akt signaling. Acta Pharmacol. Sin. 2019, 40, 1205–1211.
- Cao, L.; Graue-Hernandez, E.O.; Tran, V.; Reid, B.; Pu, J.; Mannis, M.J.; Zhao, M. Downregulation of PTEN at Corneal Wound Sites Accelerates Wound Healing through Increased Cell Migration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2272–2278.
- Li, J.; Qi, X.; Wang, X.; Li, W.; Li, Y.; Zhou, Q. PTEN Inhibition Facilitates Diabetic Corneal Epithelial Regeneration by Reactivating Akt Signaling Pathway. Transl. Vis. Sci. Technol. 2020, 9, 5.
- Gekka, M.; Miyata, K.; Nagai, Y.; Nemoto, S.; Sameshima, T.; Tanabe, T.; Maruoka, S.; Nakahara, M.; Kato, S.; Amano, S. Corneal epithelial barrier function in diabetic patients. Cornea 2004, 23, 35–37.
- Mansoor, H.; Tan, H.C.; Lin, M.T.Y.; Mehta, J.S.; Liu, Y.C. Diabetic Corneal Neuropathy. J. Clin. Med. 2020, 9, 3956.
- Shaheen, B.S.; Bakir, M.; Jain, S. Corneal nerves in health and disease. Surv. Ophthalmol. 2014, 59, 263–285.
- He, J.; Bazan, N.G.; Bazan, H.E.P. Mapping the entire human corneal nerve architecture. Exp. Eye Res. 2010, 91, 513–523.
- Müller, L.J.; Marfurt, C.F.; Kruse, F.; Tervo, T.M. Corneal nerves: Structure, contents and function. Exp. Eye Res. 2003, 76, 521–542.
- Müller, L.J.; Vrensen, G.F.; Pels, L.; Cardozo, B.N.; Willekens, B. Architecture of human corneal nerves. Invest. Ophthalmol. Vis. Sci. 1997, 38, 985–994.
- Müller, L.J.; Pels, L.; Vrensen, G. Ultrastructural organization of human corneal nerves. Investig. Ophthalmol. Vis. Sci. 1996, 37, 476–488.
- Dartt, D.A.; Bex, P.; D’Amore, P.; Dana, R.; Mcloon, L.; Niederkorn, J. Ocular Periphery and Disorders; Academic Press: Cambridge, MA, USA, 2011.
- Belmonte, C.; Garcia-Hirschfeld, J.; Gallar, J. Neurobiology of ocular pain. Prog. Retin. Eye Res. 1997, 16, 117–156.
- Maugeri, G.; D’Amico, A.G.; Amenta, A.; Saccone, S.; Federico, C.; Reibaldi, M.; Russo, A.; Bonfiglio, V.; Avitabile, T.; Longo, A.; et al. Protective effect of PACAP against ultraviolet B radiation-induced human corneal endothelial cell injury. Neuropeptides 2020, 79, 101978.
- Wu, M.; Hill, L.J.; Downie, L.E.; Chinnery, H.R. Neuroimmune crosstalk in the cornea: The role of immune cells in corneal nerve maintenance during homeostasis and inflammation. Prog. Retin. Eye Res. 2022, 91, 101105.
- Bikbova, G.; Oshitari, T.; Baba, T.; Yamamoto, S. Neuronal changes in the diabetic cornea: Perspectives for neuroprotection. BioMed Res. Int. 2016, 2016, 5140823.
- Tomlinson, D.; Fernyhough, P.; Diemel, L. Role of neurotrophins in diabetic neuropathy and treatment with nerve growth factors. Diabetes 1997, 46, S43–S49.
- Puri, S.; Kenyon, B.M.; Hamrah, P. Immunomodulatory Role of Neuropeptides in the Cornea. Biomedicines 2022, 10, 1985.
- Yang, L.; Di, G.; Qi, X.; Qu, M.; Wang, Y.; Duan, H.; Danielson, P.; Xie, L.; Zhou, Q. Substance P promotes diabetic corneal epithelial wound healing through molecular mechanisms mediated via the neurokinin-1 receptor. Diabetes 2014, 63, 4262–4274.
- Chikamoto, N.; Chikama, T.-i.; Yamada, N.; Nishida, T.; Ishimitsu, T.; Kamiya, A. Efficacy of substance P and insulin-like growth factor-1 peptides for preventing postsurgical superficial punctate keratopathy in diabetic patients. Jpn. J. Ophthalmol. 2009, 53, 464–469.
- Nagano, T.; Nakamura, M.; Nakata, K.; Yamaguchi, T.; Takase, K.; Okahara, A.; Ikuse, T.; Nishida, T. Effects of Substance P and IGF-1 in Corneal Epithelial Barrier Function and Wound Healing in a Rat Model of Neurotrophic Keratopathy. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3810–3815.
- Yang, L.W.Y.; Mehta, J.S.; Liu, Y.-C. Corneal neuromediator profiles following laser refractive surgery. Neural Regen. Res. 2021, 16, 2177.
- Zhang, Y.; Gao, N.; Wu, L.; Lee, P.S.; Me, R.; Dai, C.; Xie, L.; Yu, F.-S.X. Role of VIP and sonic hedgehog signaling pathways in mediating epithelial wound healing, sensory nerve regeneration, and their defects in diabetic corneas. Diabetes 2020, 69, 1549–1561.
- Dogru, M.; Katakami, C.; Inoue, M. Tear function and ocular surface changes in noninsulin-dependent diabetes mellitus. Ophthalmology 2001, 108, 586–592.
- Cousen, P.; Cackett, P.; Bennett, H.; Swa, K.; Dhillon, B. Tear production and corneal sensitivity in diabetes. J. Diabetes Its Complicat. 2007, 21, 371–373.
- Kabosova, A.; Kramerov, A.A.; Aoki, A.M.; Murphy, G.; Zieske, J.D.; Ljubimov, A.V. Human diabetic corneas preserve wound healing, basement membrane, integrin and MMP-10 differences from normal corneas in organ culture. Exp. Eye Res. 2003, 77, 211–217.
- Al-Aqaba, M.A.; Dhillon, V.K.; Mohammed, I.; Said, D.G.; Dua, H.S. Corneal nerves in health and disease. Prog. Retin. Eye Res. 2019, 73, 100762.
- Dua, H.S.; Said, D.G.; Messmer, E.M.; Rolando, M.; Benitez-del-Castillo, J.M.; Hossain, P.N.; Shortt, A.J.; Geerling, G.; Nubile, M.; Figueiredo, F.C. Neurotrophic keratopathy. Prog. Retin. Eye Res. 2018, 66, 107–131.
- Barsegian, A.; Lee, J.; Salifu, M.O.; McFarlane, S.I. Corneal neuropathy: An underrated manifestation of diabetes mellitus. J. Clin. Endocrinol. Diabetes 2018, 2.
- De Clerck, E.E.B.; Schouten, J.; Berendschot, T.; Koolschijn, R.S.; Nuijts, R.; Schram, M.T.; Schaper, N.C.; Henry, R.M.A.; Dagnelie, P.C.; Ruggeri, A.; et al. Reduced corneal nerve fibre length in prediabetes and type 2 diabetes: The Maastricht Study. Acta Ophthalmol. 2020, 98, 485–491.
- Zhou, T.; Lee, A.; Lo, A.C.Y.; Kwok, J. Diabetic Corneal Neuropathy: Pathogenic Mechanisms and Therapeutic Strategies. Front. Pharmacol. 2022, 13, 816062.
- Chen, X.; Graham, J.; Petropoulos, I.N.; Ponirakis, G.; Asghar, O.; Alam, U.; Marshall, A.; Ferdousi, M.; Azmi, S.; Efron, N. Corneal nerve fractal dimension: A novel corneal nerve metric for the diagnosis of diabetic sensorimotor polyneuropathy. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1113–1118.
- Li, Q.; Zhong, Y.; Zhang, T.; Zhang, R.; Zhang, Q.; Zheng, H.; Ji, L.; Sun, W.; Zhu, X.; Zhang, S. Quantitative analysis of corneal nerve fibers in type 2 diabetics with and without diabetic peripheral neuropathy: Comparison of manual and automated assessments. Diabetes Res. Clin. Pract. 2019, 151, 33–38.
- Lewis, E.J.; Lovblom, L.E.; Ferdousi, M.; Halpern, E.M.; Jeziorska, M.; Pacaud, D.; Pritchard, N.; Dehghani, C.; Edwards, K.; Srinivasan, S. Rapid corneal nerve fiber loss: A marker of diabetic neuropathy onset and progression. Diabetes Care 2020, 43, 1829–1835.
- Sady, C.; Khosrof, S.; Nagaraj, R. Advanced Maillard reaction and crosslinking of corneal collagen in diabetes. Biochem. Biophys. Res. Commun. 1995, 214, 793–797.
- Mocan, M.C.; Durukan, I.; Irkec, M.; Orhan, M. Morphologic alterations of both the stromal and subbasal nerves in the corneas of patients with diabetes. Cornea 2006, 25, 769–773.
- Saghizadeh, M.; Brown, D.J.; Castellon, R.; Chwa, M.; Huang, G.H.; Ljubimova, J.Y.; Rosenberg, S.; Spirin, K.S.; Stolitenko, R.B.; Adachi, W. Overexpression of matrix metalloproteinase-10 and matrix metalloproteinase-3 in human diabetic corneas: A possible mechanism of basement membrane and integrin alterations. Am. J. Pathol. 2001, 158, 723–734.
- Gül, M.; Emre, S.; Esrefoglu, M.; Vard, N. Protective effects of melatonin and aminoguanidine on the cornea in streptozotocin-induced diabetic rats. Cornea 2008, 27, 795–801.
- Leppin, K.; Behrendt, A.K.; Reichard, M.; Stachs, O.; Guthoff, R.F.; Baltrusch, S.; Eule, J.C.; Vollmar, B. Diabetes mellitus leads to accumulation of dendritic cells and nerve fiber damage of the subbasal nerve plexus in the cornea. Invest. Ophthalmol. Vis. Sci. 2014, 55, 3603–3615.
- Sierra-Silvestre, E.; Andrade, R.J.; Holguín-Colorado, L.; Edwards, K.; Coppieters, M.W. Occurrence of corneal sub-epithelial microneuromas and axonal swelling in people with diabetes with and without (painful) diabetic neuropathy. Diabetologia 2023, 66, 1719–1734.
- De Cilla, S.; Ranno, S.; Carini, E.; Fogagnolo, P.; Ceresara, G.; Orzalesi, N.; Rossetti, L.M. Corneal subbasal nerves changes in patients with diabetic retinopathy: An in vivo confocal study. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5155–5158.
- He, J.; Bazan, H.E. Mapping the nerve architecture of diabetic human corneas. Ophthalmology 2012, 119, 956–964.
- Davidson, E.P.; Coppey, L.J.; Kardon, R.H.; Yorek, M.A. Differences and similarities in development of corneal nerve damage and peripheral neuropathy and in diet-induced obesity and type 2 diabetic rats. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1222–1230.
- Wang, F.; Gao, N.; Yin, J.; Fu-Shin, X.Y. Reduced innervation and delayed re-innervation after epithelial wounding in type 2 diabetic Goto-Kakizaki rats. Am. J. Pathol. 2012, 181, 2058–2066.
- Stem, M.S.; Hussain, M.; Lentz, S.I.; Raval, N.; Gardner, T.W.; Pop-Busui, R.; Shtein, R.M. Differential reduction in corneal nerve fiber length in patients with type 1 or type 2 diabetes mellitus. J. Diabetes Its Complicat. 2014, 28, 658–661.
- Zhivov, A.; Winter, K.; Hovakimyan, M.; Peschel, S.; Harder, V.; Schober, H.-C.; Kundt, G.; Baltrusch, S.; Guthoff, R.F.; Stachs, O. Imaging and quantification of subbasal nerve plexus in healthy volunteers and diabetic patients with or without retinopathy. PLoS ONE 2013, 8, e52157.
- Kalteniece, A.; Ferdousi, M.; Azmi, S.; Marshall, A.; Soran, H.; Malik, R.A. Keratocyte density is reduced and related to corneal nerve damage in diabetic neuropathy. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3584–3590.
- Ferdousi, M.; Romanchuk, K.; Mah, J.K.; Virtanen, H.; Millar, C.; Malik, R.A.; Pacaud, D. Early corneal nerve fibre damage and increased Langerhans cell density in children with type 1 diabetes mellitus. Sci. Rep. 2019, 9, 8758.
- Qu, J.-h.; Li, L.; Tian, L.; Zhang, X.-y.; Thomas, R.; Sun, X.-G. Epithelial changes with corneal punctate epitheliopathy in type 2 diabetes mellitus and their correlation with time to healing. BMC Ophthalmol. 2018, 18, 1.
- Lagali, N.S.; Allgeier, S.; Guimaraes, P.; Badian, R.A.; Ruggeri, A.; Köhler, B.; Utheim, T.P.; Peebo, B.; Peterson, M.; Dahlin, L.B. Reduced corneal nerve fiber density in type 2 diabetes by wide-area mosaic analysis. Investig. Ophthalmol. Vis. Sci. 2017, 58, 6318–6327.
- Issar, T.; Tummanapalli, S.S.; Kwai, N.C.G.; Chiang, J.C.B.; Arnold, R.; Poynten, A.M.; Markoulli, M.; Krishnan, A.V. Associations between acute glucose control and peripheral nerve structure and function in type 1 diabetes. Diabet. Med. 2020, 37, 1553–1560.
- Vincent, A.M.; Edwards, J.L.; McLean, L.L.; Hong, Y.; Cerri, F.; Lopez, I.; Quattrini, A.; Feldman, E.L. Mitochondrial biogenesis and fission in axons in cell culture and animal models of diabetic neuropathy. Acta Neuropathol. 2010, 120, 477–489.
- Hamid, H.S.; Mervak, C.M.; Münch, A.E.; Robell, N.J.; Hayes, J.M.; Porzio, M.T.; Singleton, J.R.; Smith, A.G.; Feldman, E.L.; Lentz, S.I. Hyperglycemia-and neuropathy-induced changes in mitochondria within sensory nerves. Ann. Clin. Transl. Neurol. 2014, 1, 799–812.
- Vincent, A.M.; Mclean, L.L.; Backus, C.; Feldman, E.L. Short-term hyperglycemia produces oxidative damage and apoptosis in neurons. FASEB J. 2005, 19, 1–24.
- Ishibashi, F.; Kojima, R.; Taniguchi, M.; Kosaka, A.; Uetake, H.; Tavakoli, M. The expanded bead size of corneal C-nerve fibers visualized by corneal confocal microscopy is associated with slow conduction velocity of the peripheral nerves in patients with type 2 diabetes mellitus. J. Diabetes Res. 2016, 2016, 3653459.
- Zherebitskaya, E.; Akude, E.; Smith, D.R.; Fernyhough, P. Development of selective axonopathy in adult sensory neurons isolated from diabetic rats: Role of glucose-induced oxidative stress. Diabetes 2009, 58, 1356–1364.
- Ryle, C.; Donaghy, M. Non-enzymatic glycation of peripheral nerve proteins in human diabetics. J. Neurol. Sci. 1995, 129, 62–68.
- Sugimoto, K.; Yasujima, M.; Yagihashi, S. Role of advanced glycation end products in diabetic neuropathy. Curr. Pharm. Des. 2008, 14, 953–961.
- Sugimoto, K.; Nishizawa, Y.; Horiuchi, S.; Yagihashi, S. Localization in human diabetic peripheral nerve of Nɛ-carboxymethyllysine-protein adducts, an advanced glycation endproduct. Diabetologia 1997, 40, 1380–1387.
- Kaji, Y.; Amano, S.; Usui, T.; Suzuki, K.; Tanaka, S.; Oshika, T.; Nagai, R.; Horiuchi, S. Advanced glycation end products in Descemet’s membrane and their effect on corneal endothelial cell. Curr. Eye Res. 2001, 23, 469–477.
- Lyu, Y.; Zeng, X.; Li, F.; Zhao, S. The effect of the duration of diabetes on dry eye and corneal nerves. Contact Lens Anterior Eye 2019, 42, 380–385.
- Greene, D.A.; Sima, A.A.; Stevens, M.J.; Feldman, E.L.; Lattimer, S.A. Complications: Neuropathy, pathogenetic considerations. Diabetes Care 1992, 15, 1902–1925.
- Lehning, E.J.; LoPachin, R.M.; Mathew, J.; Eichberg, J. Changes in Na-K ATPase and protein kinase C activities in peripheral nerve of acrylamide-treated rats. J. Toxicol. Environ. Health Part. A Curr. Issues 1994, 42, 331–342.
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 2010, 106, 1319–1331.
- Rieger, G. The importance of the precorneal tear film for the quality of optical imaging. Br. J. Ophthalmol. 1992, 76, 157–158.
- Yang, C.-J.; Anand, A.; Huang, C.-C.; Lai, J.-Y. Unveiling the Power of Gabapentin-Loaded Nanoceria with Multiple Therapeutic Capabilities for the Treatment of Dry Eye Disease. ACS Nano 2023, 17, 25118–25135.
- Imam, S.; Elagin, R.B.; Jaume, J.C. Diabetes-associated dry eye syndrome in a new humanized transgenic model of type 1 diabetes. Mol. Vis. 2013, 19, 1259–1267.
- Zhang, X.; Zhao, L.; Deng, S.; Sun, X.; Wang, N. Dry Eye Syndrome in Patients with Diabetes Mellitus: Prevalence, Etiology, and Clinical Characteristics. J. Ophthalmol. 2016, 2016, 8201053.
- Research in Dry Eye: Report of the Research Subcommittee of the International Dry Eye WorkShop (2007). Ocul. Surf. 2007, 5, 179–193.
- Kesarwani, D.; Rizvi, S.W.A.; Khan, A.A.; Amitava, A.K.; Vasenwala, S.M.; Siddiqui, Z. Tear film and ocular surface dysfunction in diabetes mellitus in an Indian population. Indian. J. Ophthalmol. 2017, 65, 301–304.
- Wu, Y.-C.; Buckner, B.R.; Zhu, M.; Cavanagh, H.D.; Robertson, D.M. Elevated IGFBP3 Levels in Diabetic Tears: A Negative Regulator of IGF-1 Signaling in the Corneal Epithelium. Ocul. Surf. 2012, 10, 100–107.
- Zhao, Z.; Liu, J.; Shi, B.; He, S.; Yao, X.; Willcox, M.D. Advanced glycation end product (AGE) modified proteins in tears of diabetic patients. Mol. Vis. 2010, 16, 1576–1584.
- Liu, H.; Sheng, M.; Liu, Y.; Wang, P.; Chen, Y.; Chen, L.; Wang, W.; Li, B. Expression of SIRT1 and oxidative stress in diabetic dry eye. Int. J. Clin. Exp. Pathol. 2015, 8, 7644–7653.
- Wang, B.; Zeng, H.; Zuo, X.; Yang, X.; Wang, X.; He, D.; Yuan, J. TLR4-Dependent DUOX2 Activation Triggered Oxidative Stress and Promoted HMGB1 Release in Dry Eye. Front. Med. 2021, 8, 781616.
- Qu, M.; Wan, L.; Dong, M.; Wang, Y.; Xie, L.; Zhou, Q. Hyperglycemia-induced severe mitochondrial bioenergetic deficit of lacrimal gland contributes to the early onset of dry eye in diabetic mice. Free Radic. Biol. Med. 2021, 166, 313–323.
- Richdale, K.; Chao, C.; Hamilton, M. Eye care providers’ emerging roles in early detection of diabetes and management of diabetic changes to the ocular surface: A review. BMJ Open Diabetes Res. Care 2020, 8, e001094.