Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Cancer cells, especially cancer stem cells (CSCs), share many molecular features with induced pluripotent stem cells (iPSCs) that enable the derivation of induced pluripotent cancer cells by reprogramming malignant cells. Conversely, normal iPSCs can be converted into cancer stem-like cells with the help of tumor microenvironment components and genetic manipulation. These CSC models can be utilized in oncogenic initiation and progression studies, understanding drug resistance, and developing novel therapeutic strategies.
- induced pluripotent stem cells
- cancer stem cells
- reprogramming
- pluripotency
- interconversion
- cancer stem cell models
- induced pluripotent cancer cells
1. Shared Molecular Features between iPSCs and Cancer Cells
The application of iPSC technology in cancer research benefits from a high extent of shared molecular features between iPSCs and cancer cells (Figure 1A). This section explores the parallel molecular signatures and signaling pathways commonly manifested between iPSCs and cancer cells.
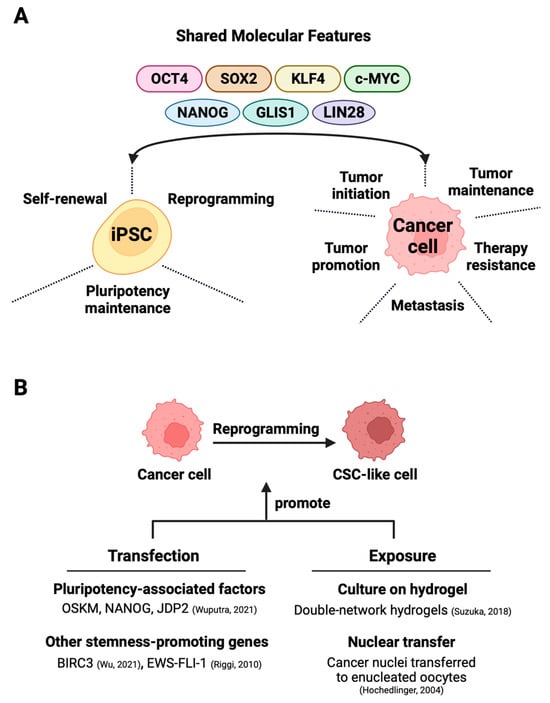
Figure 1. Exploiting shared molecular characteristics between iPSCs and cancer cells for the generation of cancer stem cells. (A) The shared factors not only support the self-renewal, pluripotency, and maintenance of iPSCs but also partake in oncogenesis in various cancers. (B) The direct reprogramming of bulk cancer cells into cancer stem cells can occur through two main methods—one involves techniques like that of transfection that induces the expression of stemness-promoting proteins [25,26] or triggers factor-dependent reprogramming [27]. The alternative exposure-based method relies on hydrogel-activated reprogramming [28] or nuclear transfer techniques [29] to drive CSC formation.
1.1. The Role of the Four De Facto Pluripotency Inducers in Cancer Cells
Octamer-binding transcription factor 4 (Oct4), also known as Oct3 or POU5F1 (POU domain, class 5, transcription factor 1), is a prominent regulator of the induction and maintenance of cellular pluripotency and is capable of reprogramming neural stem cells to pluripotency individually [30]. Oct4 overexpression, although circumstantial, has been linked to tumorigenesis in gastric cancer cells [31] and lung adenocarcinoma [32]. Additionally, Oct4 has been observed to play a role in the maintenance and progression of tumors in breast cancer [33], nasopharyngeal carcinoma [34], bladder cancer [35], rectal cancer [36], brain cancer [37], and ovarian cancer [38] as well as in chemoresistance in bladder cancer [39]. Oct4 predominantly exerts its functions through the formation of complexes with other proteins, such as Sox2, Nanog, beta-catenin, and others. Given its participation in multiple pathways associated with tumorigenesis and tumor maintenance, Oct4 may emerge as a promising target for cancer treatment strategies [40].
SRY (sex-determining region Y)-box 2, also known as Sox2, is another crucial factor for pluripotency induction in human somatic cells, and its cooperativity with Oct4 is instrumental for its function [41]. Sox2 overexpression has been linked to oncogenic initiation, amplification, and maintenance in ovarian [38], lung [42], and pancreatic [43] cancers. It has been observed to play a role in late carcinogenesis in prostate [44] and pancreatic cancer [45] as well as in chemoresistance in prostate cancer [44].
Transcription factor Krüppel-like factor 4 (Klf4) interacts directly with Oct4 and Sox2 to activate Nanog, which is another transcription factor that is responsible for the maintenance of pluripotent cells in the inner cell mass of blastocysts by blocking stem cell differentiation [46,47]. Unlike the other pluripotency genes, Klf4 has been observed to possess a tumor-suppressive role in gastric [48] and colorectal [49] cancers. However, its oncogenic role is indicated in the cases of breast cancer [50] and squamous cell carcinoma of the esophagus [51]. The diverse functions exhibited by Klf4 likely stem from its distinctive structure that encompasses both transcriptional activation and repression domains. Furthermore, numerous proteins associated with tumorigenesis, such as p21, p27, p53, and Cyclin D, are downstream targets of Klf4. Its involvement in inflammation and precancerous lesions adds to its significance in tumorigenesis studies [52].
Transcription factor c-Myc enhances pluripotency to generate high-quality iPSCs along with the previously mentioned factors [53]. Overexpression of c-Myc has been tied to prostatic neoplasia [54], and the inactivation of its antagonist tumor suppressors BRCA1 and SMARCB1 has been observed to result in its overexpression in breast cancer [55] and malignant brain rhabdoid tumors [56], respectively. Conversely, c-Myc inactivation has been shown to cause the regression of liver tumors, corroborating its role as an oncogene [57].
1.2. The Role of Auxiliary Pluripotency-Related Factors in Cancer Cells
Despite being related to Klf4, which has known tumor-suppressive properties, Nanog has been linked to the promotion of tumor growth in breast cancer [58] as well as chemoresistance and regenerative capacity in prostate cancer [59]. It has also been observed to be overexpressed in hypoxia-induced aggressiveness of prostatic and pancreatic cancers [60,61].
Glis family zinc finger 1 (Glis1) is a pro-reprogramming factor that acts in collaboration with the main pluripotency inducers [62,63]. Overexpression of Glis1 has been reported in breast cancer cells where it enhances cell migration and invasion capacity together with CUX1 [64]. A similar role of the protein is observed in ovarian cancer cells as well [65].
LIN28a/b (LIN28) is an RNA-binding protein that regulates development-associated genes post-transcriptionally [66]. It has been shown to enhance reprogramming efficiency [67] and has been linked to the aggressiveness of esophageal cancer [68], the initiation and maintenance of liver cancer [69], chemoresistance in breast cancer [70], and the suppression of the p53 tumor suppressor gene [71].
2. Cancer Stem Cells
Cancer stem cells (CSCs) constitute a small subset of cells within tumors, demonstrating the capacity for self-renewal, differentiation, and the initiation of tumor formation upon transplantation into an animal host [22]. They also display a higher expression of drug efflux pumps and elevated DNA repair activity [72]. These properties position CSCs as the primary contributors to resistance against chemotherapy and radiotherapy as well as the recurrence and relapse of cancer [22,72].
2.1. Theories of Cancer Stem Cell Origin
The origin of the CSC concept can be traced back to 1875 when Julius Cohnheim et al. proposed the “embryonal rest” theory, which suggests that cancerous growth may arise from residual embryonic cells persisting after development and remaining dormant until activation [73,74,75]. The initial modern identification of CSCs occurred through a CD34+/CD38− subpopulation of malignant cells being isolated from human acute myeloid leukemia (AML) [76]. This specific cell type demonstrated the ability to generate colony-forming progenitors when engrafted into SCID mice. Over time, various CSC biomarkers have been identified for different cancers, including breast, prostate, brain, stomach, liver, and others [77].
CSC genesis within tumors remains a point of contention. The “progenitor origin model” postulates that CSCs emerge from adult stem or progenitor cells undergoing carcinogenesis due to accumulated mutations [78], with some retaining stemness characteristics while others differentiate. This model complies with the “hierarchy model”, which states that tumors comprise a large number of differentiated or differentiating bulk cancer cells without proliferative capacity and a small population of CSCs that give rise to the bulk cells through division and subsequent differentiation [79]. The progenitor hypothesis identifies CSCs as the cells of cancer’s origin, but some argue that CSCs are instead “cancer-propagating” cells rather than “cancer-initiating” cells in the original tumor [12]. Alternatively, the “stochastic model” of CSC origin suggests that CSCs arise de novo from any cancer cell in the tumor under appropriate microenvironmental cues [79]. The hierarchical and stochastic models are successful in explaining the characteristic tumor architectures of disparate cancers [76,80,81]; their unification is as yet elusive in the context of CSC origin.
2.2. Cellular Plasticity in Cancer Cells
Cancer cell plasticity is a fundamental concept of the stochastic model of CSC origin and enables the acquisition of stem cell features by bulk cancer cells. In 2013, Ischenko et al. showed that the conditional expression of oncogenic KrasG12D in non-stem mouse cells resulted in the emergence of stemness features and metastatic potential. Subsequent analysis revealed c-Myc as a determining factor in the transformation of KrasG12D-expressing cells [82]. Further supporting the idea, Schwitalla et al. demonstrated a positive correlation between NF-κB signaling and the Wnt pathway that drives the dedifferentiation of colon cancer cells [83]. More recently, BIRC3 overexpression has been linked to enhanced self-renewal and stemness maintenance in glioblastoma cell lines and patient-derived glioblastoma cells [25].
In addition, microenvironmental signals also contribute to the generation of CSCs. Vermeulen et al. reported the impact of myofibroblast-secreted factors, such as the hepatocyte growth factor (HGF), in amplifying Wnt signaling activity and consequently triggering the formation of CSCs in colon cancer cells [84]. In an unexpected revelation, Landsberg et al. discovered that proinflammatory cytokine tumor necrosis factor-alpha (TNF-α) could facilitate the dedifferentiation of melanoma cells, leading to resistance against cytotoxic T cells [85].
2.3. Pluripotency-Associated Genes in Cancer Stem-like Feature Acquisition
Elucidating the role of pluripotency-associated genes in CSC formation and malignant activity is critical for obtaining desirable cancer stem-like cells in vitro. In 2010, Riggi et al. discovered that the EWS-FLI-1 fusion gene, the primary driver of Ewing sarcoma, could stimulate the expression of oncogenes SOX2, OCT4, and NANOG in human pediatric mesenchymal stem cells. Among these factors, SOX2 emerged as the pivotal element, with its lone expression capable of inducing CSC features in a primary tumor [26]. Yin et al. later demonstrated that the co-expression of OCT4 and NANOG in hepatocellular carcinoma (HCC) accelerated the epithelial–mesenchymal transition (EMT) process through the STAT3/Snail signaling pathway. The transfected HCC cells acquired CSC traits, including self-renewal, drug resistance, high tumorigenicity, and increased proliferation [86].
Recently, Liu et al. showed that biomechanical forces facilitate the interaction between TAZ and NANOG leading to the upregulation of SOX2 and OCT4, thereby enhancing CSC properties in human breast cancer [87]. Qi et al. previously identified a strong association between KLF4 and the stemness of human osteosarcoma cancer cells. Overexpression of KLF4 resulted in an increased sphere-forming potential, elevated expression of stemness genes, and heightened metastatic potential, with the p38 MAPK pathway implicated in the cell transition [88]. Kim et al. demonstrated that c-MYC could promote stemness and tumorigenicity in triple-negative breast cancer by inhibiting tumor suppressor zinc finger transcription factor 148 (ZNF148) [89]. Additionally, high c-MYC activity was observed in CD133+ colon CSCs, and the knockdown of c-MYC significantly attenuated CSC properties both in vitro and in vivo [90].
3. Deriving Cancer Stem-like Cells from iPSCs
Conversion of iPSCs into cancer stem-like cells can be achieved through the genetic manipulation of the iPSCs [14,91]. Given the lethal effect of some oncogenic mutations on iPSCs [91,92], inducible gene expression systems are widely used to obtain progenitor cells with CSC characteristics. Such systems are extremely useful for studying the stage- and tissue-specific oncogenicity of cancer-predisposing mutations in early carcinogenesis events. However, as previously discussed, the cell origin of cancer may not be identical to cancer-propagating or cancer-initiating CSCs [12]. Therefore, iPSCs with cancer-causing mutations are more useful for cancer initiation studies than CSC modeling. Yet, a handful of studies, mostly focusing on brain tumor modeling, reported cancer stem cell-like properties in engineered/edited iPSC-derived stem and progenitor cell types. Haag et al. showed that the H3.3-K27M mutation in iPSC-derived neural stem cells led to stemness maintenance and increased proliferation through the gliomagenic cells [91], while an earlier study by Koga et al. demonstrated the high self-renewal capacity of PTEN−/−; NF1−/− and TP53−/−; PDGFRAΔ8–9 iPSC-derived high-grade glioma (HGG) spheres [14]. In 2012, Friedmann-Morvinski et al. conducted a study on mouse models where they introduced a lentiviral construct carrying two shRNA sequences against NF1 and TP53 genes into astrocytes and neurons to dedifferentiate the cells into glioblastoma multiforme (GBM)-forming neural progenitor/stem-like cells [93]. Though not relating to iPSCs, this study provides strong evidence for CSC induction through the conditional manipulation of gene expression even in differentiated cells.
Exposure of normal stem cells to malignant niches has been proposed as a model for CSC origin [94,95,96]. Conforming to this hypothesis, CSC models were generated in vitro either by exposing normal iPSCs to conditioned media (CM) from cancer cells of different tissue origin [13,97,98,99,100] or by coculturing the iPSCs with the cancer cells [13] (Figure 2A). A seminal study involving various mouse cancer cell lines demonstrated that CM-dependent CSC derivation from mouse iPSCs (miPSCs) was more successful than the coculture method [13]. The CM-exposed miPSCs generally exhibited CSC-like properties by manifesting self-renewal in sphere-formation assays and forming tumors when transplanted into nude mice [13,97,98,99,100]. A noteworthy observation is that tumors derived from different CM-based CSCs exhibit varying angiogenic and metastatic potential apparently due to the difference in the expression of metastasis and angiogenesis regulators, such as IFNγ and MMPs, in corresponding CSCs [13].
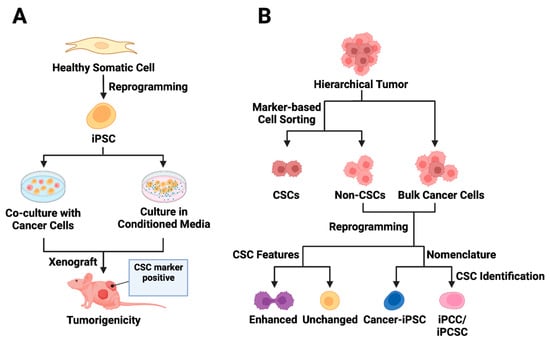
Figure 2. Utilizing iPSC technology in cancer stem cell modeling. (A) Exposure-based approaches to CSC model derivation from iPSCs are presented. The first approach involves co-culturing normal iPSCs with cancer cells, while the second strategy entails culturing iPSCs in conditioned media from cancer cells. The iPSCs exposed to tumor microenvironment factors acquire distinct CSC features and demonstrate the ability to form tumors in vivo. (B) The reprogramming of cancer cells is depicted, acknowledging the hierarchical structure of tumors. Cell sorting enables the separation of CSC marker-expressing cells from bulk tumor cells, though the presence of CSC markers may not always correlate with CSC features (non-CSCs). Following the reprogramming of non-stem cancer cells, some cancer-derived iPSCs (cancer-iPSCs) may exhibit enhanced CSC features. The reprogrammed cell population is called cancer-iPSCs, induced pluripotent cancer cells (iPCCs), or induced pluripotent cancer stem cells (iPCSCs).
CM-based CSCs can be differentiated into various stromal cells that bolster tumor growth. Subpopulations of CM CSCs have been shown to express VEGF-A [97,99] and FGF2 [99] angiogenic factors in vitro and in vivo to mediate the differentiation and maturation of CSC-derived endothelial cells through paracrine signaling [99]. Furthermore, iPSC-derived CSC-like cells have also been reported to give rise to cancer-associated fibroblasts [100], supporting tumor maintenance and CSC survival [101]. Tumor-associated myoepithelial cells can also arise from these models upon mammary fat pad transplantation [102].
The long-term tumorigenicity of these CSC models remains largely uncharacterized. However, serial transplantation of CSCs induced using pancreatic carcinoma-conditioned media into mice was shown to form more aggressive cancers with the downregulation of the CSC markers of CD133, CD24a, and EpCAM [98]. miPSCs exposed to Lewis lung carcinoma (LLC)-CM were shown to harbor a hypomethylated genome with an enhanced PI3K–Akt pathway [103] that was retained by subsequent progenies. It is important to note that the PI3K–Akt pathway has been reported as highly necessary for the viability of human iPSCs [104]. Later analysis uncovered prostaglandin E2 (PGE2) enrichment in LLC-CM and a higher conversion of miPSCs into CSCs with PGE2 as an additive in the culture media [105]. In addition, extracellular vesicles from LLC cells have been shown to render miPSCs capable of forming tumors in vivo [106]. Exposure to the E-cadherin-Fc chimera protein was also shown to promote CSC features in colon cancer cells [107].
A higher degree of plasticity distinguishes cancer stem cells from bulk cancer cells [108], and the distinction is particularly relevant in the context of the CM-dependent generation of malignant cells from iPSCs. A recent study on the U87MG glioblastoma cell line revealed that CM from bulk cancer cells and cancer stem cell-generated neuron-like glioblastoma cells and spherical glioblastoma stem cells from iPSCs, respectively. While both the induced cell types showed the MGMT, GLI2, LEF1, and β-catenin overexpression characteristic of glioblastoma, only the cancer stem-like cells expressed stem cell markers CD133 and CD44. This finding emphasizes CM compositional variation as a critical determiner of the molecular features of the induced cells [109].
An expedited in vivo approach to converting miPSCs into CSCs entails mixing cancer cells with iPSCs before injecting them into immunocompromised mice. Pancreatic cancer stem cells generated through this method showed varied morphology (mesenchymal-like vs. epithelial-like), tumorigenicity, differentiation capacity, drug sensitivity, and gene expression depending on the distinct microenvironment provided by different cancer cell-CM. However, generated CSCs usually exhibit enhanced invasion ability as well as upregulated energy production and cancer-related pathways compared with the parental iPSC lines [110].
Finally, the differentiation of iPSCs into cervical reserve cells using small molecules has been described. These reserve cells possibly give rise to cervical cancer stem cells [111].
Tumorigenicity and Tumor-Promoting Potential of Other iPSC-Derived Cells
In an in vitro pancreatic ductal adenocarcinoma (PDAC) model, iPSC-derived stellate cells were cocultured with patient-derived organoids or cancer cells to mimic tumor stroma. The stellate cells promoted the growth of some organoids and cells but not all. The observation suggests that the growth of cancer cells depends on the properties of the original tumor and is differentially influenced by a given stromal cell type [112].
A wealth of reports has described the generation, application, and overall usability of iPSC-derived mesenchymal stem cells (iMSCs) for tissue regeneration, cancer therapy, and the treatment of immune-related diseases [113,114,115,116,117]. However, the oncogenicity of iMSCs, especially iMSCs derived from mutation-carrying iPSCs, requires careful investigation. A study showed that iMSCs derived from BRCA+/− iPSCs showed elevated expressions of VEGF, PDGF, and ANGPT angiogenic factors and promoted the formation of an extended vascular network both in vitro and in vivo. The haploinsufficient mesenchymal stem cells were also characterized by higher migration ability and significantly upregulated Periostin expression compared with its BRCA+/+ counterpart. An enhanced tumorigenic and metastatic potential of BRCA+/− iMSCs was also observed when co-injected with 4T1 breast cancer cells into the mammary tissue of NOD-SCID mice [17]. Promisingly, iMSCs derived from disease-free iPSCs are less protumorigenic, while bone marrow-derived mesenchymal stem cells tend to support tumor growth and invasion by producing PGE2, IL6, and other protumor factors [118,119]. It will be worthwhile to examine the basis of the dissimilar cancer-promoting properties between iMSCs and mesenchymal stem cells from other sources [120,121,122].
4. Reprogramming Cancer Cells to Obtain Cancer Stem-like Cells
With the advent of iPSC technology, it has been possible to convert patient-derived somatic cells with cancer-predisposing germline mutation into iPSCs for disease modeling and therapy evaluation [123,124,125]. Beyond these goals, directed dedifferentiation of malignant cells can yield CSC-like cells (Figure 2B) for cancer research. The direct reprogramming of non-stem bulk cancer cells into CSC-like cells can occur through the transfection of either CSC-promoting protein-coding genes [26,82] or pluripotency-associated genes, which is discussed in great detail in this section. An alternative method for direct reprogramming utilizes exposure-based approaches to drive CSC formation by cancer cells (Figure 1B).
Relying on reprogramming factors to induce pluripotency, an attempt to reprogram colorectal cancer cell lines with the Yamanaka cocktail yielded cancer-iPSCs that could differentiate into three germ layer lineages. Nevertheless, the down-regulation of pluripotency genes and a distinct miRNA profile suggest incomplete or partial reprogramming toward pluripotency. Additionally, the cancer-iPSCs attained an epithelial/mesenchymal hybrid phenotype owing to dysregulated miRNA expression [126].
Cancer cell reprogramming has proved an effective tool in translational medical research on myeloproliferative disorders. iPSCs derived from malignant cells of juvenile myelomonocytic leukemia (JMML) through lentiviral OSKM expression showed higher proliferative capacity in cultures and produced myeloid cells of pathological features upon differentiation [20]. More JMML models have been established after with the help of iPSC technology to facilitate the study of disease mechanisms and potent therapeutics [127,128]. iPSCs generated from imatinib-sensitive chronic myelogenous leukemia (CML) patients showed imatinib insensitivity despite restored the expression of the BCR-ABL oncoprotein. Hematopoietic differentiation of the CML-iPSCs regained sensitivity to imatinib, though a fraction of immature cells were still resistant to kinase inhibitors like CML-iPSCs [129]. Similar results were obtained for KBM7 leukemia cell line-derived iPSCs [130]. Recently, a streamlined OSKM-based reprogramming method has been described to derive iPSCs from genetically diverse AML patients, which can model the disease reliably upon xenotransplantation [131]. AML-iPSCs have previously been shown to lose the epigenetic memory of parental cells and lack oncogenic potential. However, the hematopoietic differentiation of these cells reinstates leukemic properties [132]. Cancer-iPSCs that retain oncogenic mutations and malignant properties of source cells thus present a robust and renewable source of in vitro models for drug screening and mechanistic studies of hematological malignancies [20,133,134,135,136,137].
Toward the derivation of brain CSC models, pluripotency induction using OCT4 and JDP2 reprogramming factors conferred DAOY medulloblastoma cell lines with higher in vivo tumorigenicity [27]. The enhancement of tumor formation potency may partly be explained by mTOR activation through epigenetic rewiring due to increased Oct4 [138]. Moreover, a study on DAOY, D341, and D283 medulloblastoma cell lines characterized undifferentiated cell subpopulations in each cell line phenotypically and functionally to conclude that D283 has the highest level of CSC enrichment among the medulloblastoma cell lines [139]. Genetic alterations and the position of the cell of tumor origin within the differentiation hierarchy may account for the observed heterogeneity of stemness features in cancer cell lines [140].
This entry is adapted from the peer-reviewed paper 10.3390/cells13020125
This entry is offline, you can click here to edit this entry!