1. Introduction
Corneal alterations in diabetes manifest through increased corneal thickness, epithelial defects, fragility, superficial punctate keratitis, delayed and incomplete wound repair, as well as detectable endothelial changes and neuropathy characterized by decreased corneal sensitivity [
20]. In fact, peripheral diabetic neuropathy contributes to diabetic keratopathy by damaging corneal nerve fibers due to hyperglycemia. This results in a significant reduction and alteration in subepithelial nerve fiber morphology, leading to reduced corneal surface sensitivity, a main characteristic of diabetic keratopathy [
21,
22]. Progressive corneal epithelial cell loss, recurrent erosions, ulcers, and scarring events may culminate in devastating corneal opacities, ultimately causing severe visual impairment or blindness [
23,
24].
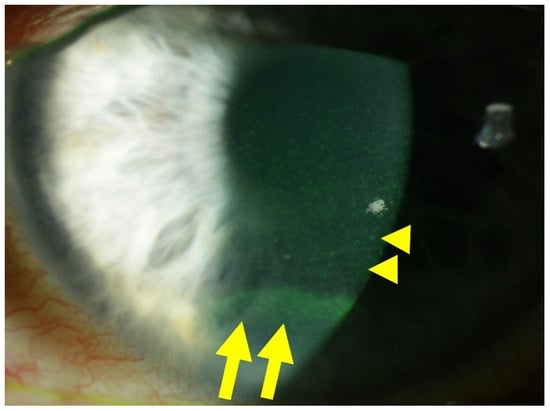
Figure 1 illustrates an example of diabetic keratopathy.
Figure 1. Slit lamp photograph of a patient with diabetic keratopathy, suffering from dry eye symptoms and recurrent corneal erosions. Fluorescein staining of the ocular surface reveals fine punctate keratitis (yellow dots), which is typical of dry eye disease (DED), highlighted by yellow arrowheads. In the inferior third of the cornea, an epithelial ridge (yellow horizontal line) is visible, indicative of a recently closed erosion, highlighted by yellow arrows.
2. Corneal Cell Damage in Diabetic Keratopathy
2.1. Redox Signaling in the Diabetic Cornea
Hyperglycemia, the primary contributor to the pathogenesis of DM and its complications, notably affects the corneal epithelium, a crucial tissue involved in diabetic keratopathy. The corneal epithelium, being a major player in this condition, exhibits insulin-independent glucose uptake through glucose transporter-1 (GLUT1), facilitating a continuous influx of glucose into cells [
25,
26,
27]. In instances of heightened metabolic demand, such as during corneal damage, the expression of GLUT1 transporters increases [
28]. Furthermore, the corneal epithelium directly receives glucose through transcorneal transport from the aqueous humor, rather than from tears, thereby maintaining high glycogen levels. During DM, the corneal epithelium becomes exposed to high glucose levels [
29]. Notably, hyperglycemia triggers an escalation in the production of advanced glycation end products (AGEs) and significantly elevates the levels of ROS [
30,
31,
32,
33,
34]. Studies by Kim and colleagues revealed that in diabetic corneal cells, the accumulation of AGEs is linked to oxidative DNA damage, measured through 8′-hydroxy-2′-deoxyguanosine levels. This cascade leads to apoptotic activation via the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), contributing to delayed epithelial wound healing in the diabetic cornea [
35]. AGEs, generated under hyperglycemic conditions, promote ROS generation by binding to their receptor RAGE (receptor of AGE). This process stimulates enhanced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX)-related ROS formation and increased phosphorylation of p47
phox. This, in turn, activates the c-Jun N-terminal kinase and p38 mitogen-activated protein kinase pathways, culminating in apoptosis and microvascular complications [
36,
37,
38,
39]. Additionally, hyperglycemia increases intracellular concentrations of diacylglycerol (DAG), activating protein kinase C (PKC) and subsequently NOX subtypes (NOX1, NOX2, and NOX4), collectively amplifying ROS formation [
38,
40,
41,
42,
43]. Furthermore, high glucose levels inhibit the epidermal growth factor receptor (EGFR)–phosphatidylinositol 3-kinase (PI3K)/Ak strain transforming (Akt, also known as protein kinase B) pathway through ROS, attenuating corneal epithelial wound healing [
44].
As an additional repercussion of the oxidative stress condition, NF-kB, a crucial transcription factor that regulates immune responses and the expression of adhesion molecules, becomes activated. This activation leads to the abnormal production of proinflammatory cytokines, contributing to chronic inflammation. This establishes a positive loop between oxidative stress, inflammation, and apoptosis in diabetic keratopathy [
35]. In addition, ROS and RAGE trigger phospholipase C/PKC/extracellular signal-regulated kinase activator protein 1 signaling, promoting fibrogenesis through transforming growth factor beta (TGF-β) expression [
45]. Notably, the TGF-β1 isoform, a major subtype expressed in the cornea, induces delayed re-epithelialization and myofibroblast transformation, suggesting its inhibition as a potential strategy to promote corneal wound healing [
46]. Of note, RAGE signaling is activated not only by AGEs but also by other ligands, such as the chemokine high-mobility group box 1 protein (HMGB1), whose expression is reported to be elevated in the corneas of diabetic murine models during wound healing. Its inhibition may be a potential molecular target [
47].
2.2. Pyroptosis in the Corneal Epithelium Triggered by Hyperglycemia
ROS abundance not only triggers aberrant inflammation via NF-kB but also induces epithelial cell death via pyroptosis. Oxidative stress can activate the nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) inflammasome in corneal epithelial cells [
48]. The NLRP3 inflammasome is a cellular multiprotein complex that plays a significant role in the immune response. The protein complex is activated when cells are damaged or infected by certain stress factors such as environmental toxins, bacteria, or viruses [
49,
50,
51]. The NLRP3 inflammasome consists of the NLRP3 protein responsible for detecting harmful signals, the apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC), and the caspase-1 protein responsible for the activation of pro-inflammatory cytokines like interleukin (IL)-1ß and IL-18 [
43,
52]. When the NLRP3 inflammasome is activated, caspase-1 provides cleavage and activation of IL-1β and IL-18, as well as of gasdermin D, whose fragments generate a membrane pore, allowing proinflammatory cytokine release and membrane disruption, finally leading to pyroptosis [
53]. These pathological processes lead to cell death in epithelia and stromal keratocytes.
2.3. Involvement of Stroma, Endothelium, and Tight Junctions in Diabetic Damage
Bitirgen et al. demonstrated a reduced basal epithelial cell density, decreased anterior stromal keratocyte counts, and endothelial cell density occurring prior to diabetic retinopathy in individuals with DM type 2 [
54]. Zhang and colleagues reported a specific susceptibility of corneal endothelial cells to ultraviolet A-related oxidative damage in a diabetic murine model. This was attributed to an attenuated parkinsonism-associated deglycase 1/nuclear factor erythroid-derived 2-related factor 2 (Nrf2)/nicotinamide adenine dinucleotide (NAD) phosphate (P)H quinone oxidoreductase 1 (NQO1) pathway [
55]. Nrf2 activation is closely related to the levels of nuclear deacetylase sirtuin 1 (SIRT1) [
56], with an upregulation of SIRT1 associated with increased Nrf2 activation [
57]. Both proteins are essential antioxidative regulators, suggesting their significance as molecular targets in antioxidant research [
58].
Additionally, hyperglycemia has been reported to impair conjunctival and corneal barrier function, potentially through a loss of laminin-5, a principal component of the corneal basement membrane [
59]. Reduced expression of the tight junction component occludin was observed in the human diabetic corneal epithelium by Huang and colleagues [
60]. Furthermore, hyperglycemia delays corneal wound healing and reduces cell migration via oxidative stress, through impairing Akt signaling [
44,
61,
62]. More specifically, the phosphatase and tensin homologue protein results upregulated in damaged corneal epithelium, and its inhibition may allow an increase in the PI3K/Akt signaling, accelerating wound healing [
63,
64].
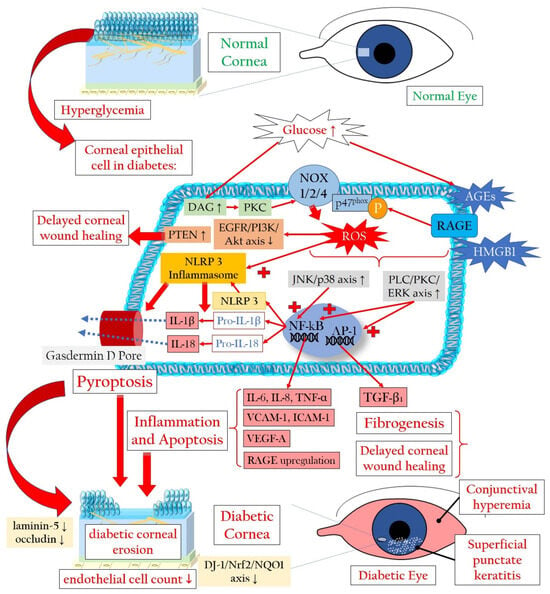
Figure 2 illustrates the mechanisms leading to a loss of corneal epithelial cells in diabetic keratopathy. The increased loss of corneal epithelial cells can lead to a reduction in the barrier function of the corneal epithelium, thereby favoring superficial punctate keratitis, recurrent erosions, persistent epithelial defects, and infections [
65].
Figure 2. Cellular signaling mechanisms that lead to the demise of corneal epithelial cells in the context of diabetic keratopathy. AGEs: advanced glycation end products; Akt: Ak strain transforming (also known as protein kinase B); DAG: diacylglycerol; IL: interleukin; DJ-1: parkinsonism-associated deglycase 1; EGFR: epidermal growth factor receptor; ERK: extracellular signal-regulated kinase; HMGB1: high-mobility group box 1 protein; ICAM-1: intercellular adhesion molecule 1; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NOX: NADPH oxidase or nicotinamide adenine dinucleotide phosphate oxidase; NQO1: NAD(P)H quinone oxidoreductase 1; Nrf2: nuclear factor erythroid-derived 2-related factor 2; P-p47phox: phosphorylated form of the protein p47phox, part of the NADPH oxidase enzyme system; PKC: protein kinase C; PI3K: phosphatidylinositol 3-kinase; PLC: phospholipase C; PTEN: phosphatase and tensin homologue protein; RAGE: receptor for advanced glycation end products; ROS: reactive oxygen species; TGF-β1: transforming growth factor beta 1; TNF-α: tumor necrosis factor alpha; VCAM-1: vascular cell adhesion molecule 1; VEGF-A: vascular endothelial growth factor A. Upward arrows indicate upregulation or increased activity, downward arrows indicate downregulation or decreased activity.
3. Diabetic Corneal Neuropathy
3.1. Evidence of Corneal Nerve Damage in Diabetes
The cornea represents the most densely innervated structure in the human body, with its nerves contributing to various homeostatic functions such as blinking movements and the release of neuropeptides and growth factors [
12,
66,
67]. The central corneal nerve density is approximately 7000 nociceptors/mm
2, making the cornea roughly 300–600 times more sensitive than the skin [
68]. Stromal nerve branches are formed by 900–1200 myelinated and unmyelinated axons with a diameter ranging between 0.5 and 5 μm [
22]. The peripheral and central nerve endings of the human cornea consist of nociceptive Aδ and C fibers [
69]. Stromal nerves are disposed parallelly to the collagen lamellae and subdivide into smaller fascicles as they extend in superficial/anterior direction, creating interconnections within the anterior stromal plexus [
67,
70]. While some stromal nerves terminate as free endings, a significant number of them directly innervate the keratocytes [
70,
71], establishing a profound and intricate relationship between corneal epithelial layers and sensory innervation [
72], particularly pertinent in cases of DM. The stroma-originated nerves give rise to the sub-basal nerve plexus (SBNP), which innervates the corneal epithelium. SBNP fibers pass through the Bowman’s layer into the epithelium, predominantly in peripheral regions. These fibers form long bundles progressing towards the center, dividing into smaller branches that interconnect, ultimately creating a complex network within the epithelium [
66].
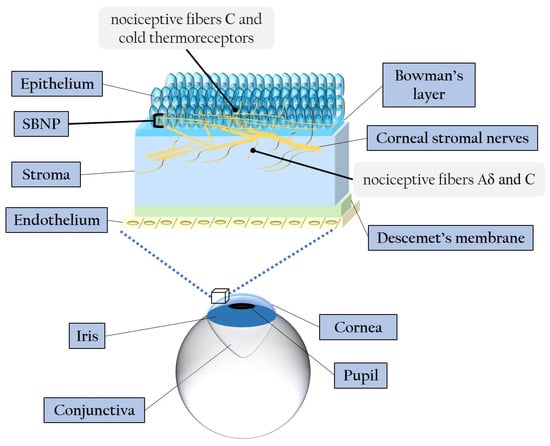
Figure 3 illustrates the corneal layers and innervation, emphasizing the distinctive distribution and ramification of stromal nerves throughout the epithelium.
Figure 3. Schematic representation of the corneal layers and innervation. SBNP: sub-basal nerve plexus.
Multiple studies have elucidated the intricate mechanisms of neuroimmune cross-talk within the cornea, primarily mediated through the interplay between neuromodulators expressed by corneal nerves and their receptors in resident corneal leukocytes [
73,
74]. Corneal sensory innervation releases neurotrophins and neuropeptides, while autonomic innervation secretes catecholamines and acetylcholine [
21,
68]. Neurotrophic molecules play a crucial role in preserving corneal homeostasis and facilitating corneal wound healing [
75]. Notably, neurotrophin-3 (NT-3) and nerve growth factor (NGF) are pivotal neurotrophic agents for ensuring corneal health and repair [
76].
The sensory nerves within the cornea produce various neuropeptides responsible for triggering diverse molecular pathways. Among these are substance P, calcitonin gene-related peptide, and α-melanocyte-stimulating hormone, which activate different molecular signaling pathways [
77]. Additionally, beyond conventional neurotransmitters, the sympathetic innervation in the cornea also involves serotonin and neuropeptide Y. Meanwhile, as regards the corneal parasympathetic innervation, activity was demonstrated for vasoactive intestinal polypeptide and galanin [
77]. Neuropeptides exert their effects by binding to receptors belonging to the superfamily of G protein-coupled receptors, thereby initiating various downstream signaling pathways [
77]. Of particular interest in managing diabetic keratopathy might be substances like substance P [
78], possibly in combination with insulin-like growth factor 1 [
79,
80], and vasoactive intestinal polypeptide [
81,
82]. These substances play roles in promoting wound healing and corneal regeneration. Delayed wound healing, reduced corneal sensitivity, and anomalies in the tear film associated with DM are intricately linked to profound injuries to corneal nerve fibers during the early phases of DM [
22,
83,
84,
85]. This correlation underscores an imbalance in the interdependence between corneal epithelium and nerves, leading to damages in both tissues, characteristic of DM-related neurotrophic keratopathy [
86,
87]. Numerous investigations have detailed critical morphological changes and a pathological decrease in SBNP fibers in DM, even during prediabetes [
88,
89,
90]. Specifically, diabetic patients exhibit reductions in nerve fiber density, branch density, and fiber length [
91,
92,
93]. Structural changes induced by DM extend to the highly innervated stromal cornea, where the accumulation of AGEs contributes to collagen crosslinking and increased corneal thickness [
94]. Moreover, abnormal increases in the tortuosity of stromal nerves have been observed in diabetic patients [
95]. Overexpression of matrix metalloproteinases (MMP)-3 and -10 in the diabetic stroma indicates significant corneal alterations and remodeling [
96]. Rodent diabetic models demonstrated stromal inflammation and edema [
97], and in diabetic murine models, dendritic cell infiltration and nerve fiber impairment in the corneal SBNP hint at a neurotrophic function of this cell subpopulation [
98]. Moreover, corneal sub-epithelial microneuromas and axonal swelling positively correlate with painful diabetic polyneuropathy, underlining the nuanced relationship between corneal nerve morphology and diabetic complications [
99].
In both experimental and clinical studies, the most significant decrease in corneal branch density and nerve fibers occurs in the SBNP, near the epithelium, emphasizing the strong association between corneal neuropathy and diabetic keratopathy [
100,
101,
102,
103,
104,
105]. Reduced corneal keratocytes in diabetic patients have been linked to damage in SBNP fibers [
106]. An increase in corneal Langerhans cells is associated with decreased SBNP fiber density [
107], while diminished keratocyte density and increased Langerhans cell density have been linked to delayed epithelial wound healing [
108]. Furthermore, the variability in serum glucose levels and the duration of DM play crucial roles in the detrimental effects on corneal nerves. For example, individuals with DM under insulin-dependent therapy for five or more years displayed diminished corneal epithelial nerve density and an abundance of nerve loops in the stroma [
101]. Longer durations of DM type 2 are positively correlated with the gradual degeneration of SBNP [
109]. Moreover, a recent study highlighted morphological changes in corneal stromal nerves in DM, underscoring an association between glucose level variability and the loss of corneal nerve length in the inferior whorl [
110].
3.2. Molecular Pathways Leading to Corneal Nerve Damage
From a molecular standpoint, hyperglycemia triggers the activation of several pathways involving mitochondria, AGEs, the polyol pathway, and the PKC cascade. These pathways collectively contribute to an excess of ROS, ultimately leading to cell death and corneal nerve injuries [
21,
90].
Hyperglycemia-Related Mitochondrial Dysfunction
In the context of DM, the influx of glucose into the mitochondria accelerates the electron transport chain, indirectly promoting the increased formation of superoxide (O
2●−), resulting in oxidative stress [
30]. Hyperglycemia induces alterations in mitochondrial dynamics, with short-term exposure leading to fission, caspase-3 activation, and oxidative stress, while prolonged exposure may trigger mitochondrial biogenesis [
115]. Chronic hyperglycemia contributes to impaired mitochondrial axonal transport due to the accumulation of larger, swollen mitochondria, resulting in decreased nerve conduction velocity, axonal demyelination, and neuron apoptosis [
116,
117,
118]. Importantly, these mitochondrial anomalies can disrupt the corneal neurotrophic interaction by decreasing the levels of NT-3 and NGF [
76,
119].
AGEs and Axonal Degeneration
During chronic hyperglycemia, AGEs not only overproduce ROS but also form cross-links with structural proteins in peripheral nerves. This process induces the formation of glycated myelin proteins, dysfunctional entities that promote demyelination, impaired regenerative activity, and axonal degeneration [
120,
121,
122]. AGEs also cross-link with collagen molecules in the corneal stroma [
94], leading to the glycation of laminin and fibronectin in Descemet’s membrane. This glycation decreases the adhesion of corneal endothelial cells [
123], potentially causing endothelial dysfunction and microangiopathy [
21].
Effects of the Polyol Pathway and PKC Cascade
The polyol pathway converts glucose into sorbitol and then into fructose, and its activation is a result of hyperglycemia-induced overload in the glycolytic pathway. This alternative metabolic pathway necessitates NADPH and NAD
+, leading to a decrease in glutathione (GSH) synthesis and subsequently hydrogen peroxide (H
2O
2) generation. As NADPH is vital for the regeneration of the antioxidant GSH, its reduced availability results in a diminished antioxidant capacity [
124]. Furthermore, an abundance of sorbitol and fructose is negatively correlated with free nerve myoinositol, a molecule crucial for normal nerve conduction velocity [
125]. The reduced bioavailability of myoinositol leads to decreased functionality of the membrane sodium–potassium adenosine triphosphatase (Na
+/K
+ ATPase), compromising nerve conduction velocity [
29].
As described above, the PKC pathway is activated in response to high glucose-induced intracellular DAG, causing enhanced ROS generation through NOX activation in corneal epithelial cells [
38,
40,
41,
42,
43]. Additionally, PKC activation can impair Na
+/K
+ ATPase function, resulting in reduced nerve conduction and regeneration [
126,
127].
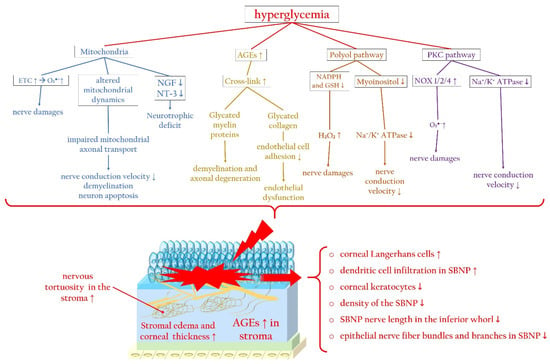
Figure 4 summarizes the main molecular pathways associated with hyperglycemia that contribute to corneal nerve damage, along with the principal changes in the corneal stroma and epithelium, focusing particularly on the SBNP during diabetic keratopathy.
Figure 4. Hyperglycemia-related molecular pathways initiating corneal nerve damage and changes in the corneal stroma and SBNP during diabetic keratopathy. Hyperglycemia causes mitochondrial dysfunction, altered mitochondrial dynamics, and neurotrophic deficits, which trigger ROS overproduction. In addition, during diabetes, the accumulation of AGEs promotes the glycation of myelin, favoring axonal degeneration, and collagen, inducing endothelial dysfunction. Furthermore, activation of the polyol and PKC signaling pathways trigger, via ROS overproduction, corneal nerve damage, impairing nerve conduction. AGEs: advanced glycated end products; ETC: electron transport chain; Na+/K+ ATPase: sodium–potassium adenosine triphosphatase; NGF: nerve growth factor; NOX: nicotinamide adenine dinucleotide phosphate oxidase; NT-3: neurotrophin-3; PKC: protein kinase C; SBNP: sub-basal nerve plexus. Upward arrows indicate upregulation or increased activity, downward arrows indicate downregulation or decreased activity.
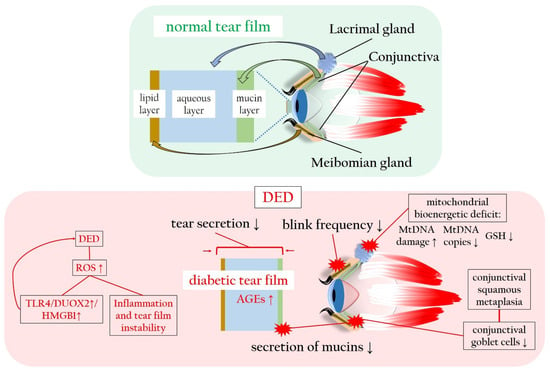
4. Diabetes-Associated Dry Eye Disease Promotes Oxidatives Stress and Inflammation
The tear function unit, encompassing cornea, conjunctiva, lacrimal gland, Meibomian glands, eyelids, and sensory/motor nerve fibers, intricately regulates tear fluid secretion and tear film formation, which are crucial for maintaining and safeguarding the tear film [
128]. Disruptions in any component of this unit can result in DED. This condition is not only linked to a deficiency in tear production, but also associated with a status of chronic inflammation and oxidative stress. In this regard, a recent innovative study by Yang and colleagues demonstrated the effectiveness of a gabapentin/nanoceria nanoformulation as a carrier with anti-inflammatory, antiangiogenic, antiapoptotic, and neuroprotective properties in a rabbit model of DED [
129].
Regarding DM, chronic hyperglycemia can potentially harm nerve fibers that supply the lacrimal gland, resulting in histological alterations within the lacrimal gland itself [
130]. DM-induced corneal nerve fiber loss diminishes the corneal sensitivity, subsequently impairing tear secretion [
12]. Moreover, patients with DM often exhibit a reduced blink frequency, a common factor in evaporative DED [
3,
131]. Conjunctival squamous metaplasia and a decline in conjunctival goblet cells contribute to decreased mucin secretion, diminishing tear film adhesion to the ocular surface and causing tear film instability [
132]. Elevated levels of insulin-like growth factor-binding protein 3 (IGFBP3) in diabetic tears act as a negative modulator of IGF-1 signaling in the corneal epithelium, exacerbating corneal impairment and DED in diabetic corneas [
133]. Furthermore, increased concentrations of AGEs in the tear fluid initiate inflammatory reactions and oxidative stress on the ocular surface, further impairing corneal epithelial cell function [
134,
135]. Notably, HMGB1, an alternative ligand of the receptor for RAGE, and dual oxidase 2 (DUOX2), a subtype of NOX, play crucial roles in DED. Hyperosmolarity-induced DUOX2 activation, mediated by toll-like receptor 4 (TLR4)-dependent signaling and exacerbated by ROS excess, leads to HMGB1 release, cell death, and inflammation in a positive loop in human corneal epithelial cells exposed to DED conditions [
136]. In a diabetic murine model, hyperglycemia induces a more severe mitochondrial bioenergetic deficiency in lacrimal gland cells compared to corneal cells. This deficiency results in elevated mtDNA damage, reduced mtDNA copies, decreased levels of GSH, contributing to the early onset of DM-induced DED [
137].
Collectively, diabetic alterations affecting tear film composition, secretion, and stability manifest as reduced tear volume and decreased break-up time and conjunctival goblet cell density. There are enhanced tear film osmolarity, squamous conjunctival metaplasia, disturbances in the ocular surface microbiota, heightening the risk of bacterial conjunctivitis, and impairment of Meibomian glands [
138].
Figure 5 illustrates the tear film sublayers and the detrimental impact of hyperglycemia and ROS on tear production and stability, leading to DED.
Figure 5. Normal and diabetic tear film in comparison, with particular focus on the ROS-related changes in tear film production, composition and stability. AGEs: advanced glycated end products; DED: dry eye disease; DUOX2: dual oxidase 2; GSH: glutathione; HMGB1: high-mobility group box 1 protein; mtDNA: mitochondrial DNA; RAGE: receptor of advanced glycated end products; ROS: reactive oxygen species; TLR4: toll-like receptor 4. Upward arrows indicate upregulation or increased activity, downward arrows indicate downregulation or decreased activity.
This entry is adapted from the peer-reviewed paper 10.3390/antiox13010120