Metabolic dysfunction-associated steatotic liver disease (MASLD) is a unique, multi-factorial condition with several phases of progression including steatosis, steatohepatitis, fibrosis, cirrhosis, and hepatocellular carcinoma. Sterol element binding protein 1c (SREBP1c) is the main transcription factor involved in regulating hepatic de novo lipogenesis. This transcription factor is synthesized as an inactive precursor, and its proteolytic maturation is initiated in the membrane of the endoplasmic reticulum upon stimulation by insulin. SREBP cleavage activating protein (SCAP) is required as a chaperon protein to escort SREBP from the endoplasmic reticulum and to facilitate the proteolytic release of the N-terminal domain of SREBP into the Golgi. SCAP inhibition prevents activation of SREBP and inhibits the expression of genes involved in triglyceride and fatty acid synthesis, resulting in the inhibition of de novo lipogenesis.
1. Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD) is an umbrella term for a broad continuum of manifestations characterized by histological findings of macrovescicular hepatic steatosis of more than 5% of the hepatocytes in individuals who consume little or no alcohol
[1]. This benign liver disease may progress to cirrhosis in more than 25% of the patients
[2]. MASLD is further divided into metabolic dysfunction-associated fatty liver (MAFL), which is characterized by steatosis with or without mild lobular inflammation, and metabolic dysfunction-associated steatohepatitis (MASH), which is characterized by diffuse lobular inflammation and fibrosis and has great potential to progress into hepatocellular carcinoma
[3]. MASLD and dyslipidemia are driven by common molecular mediators and, further, MASLD is strongly associated with dyslipidemia, representing one of the most common manifestations of the metabolic syndrome. The most important factor playing a significant role in the pathogenesis of MASLD is the crosslink between liver lipid metabolism and peripheral fat metabolism
[4].
The three sterol element binding proteins (SREBPs), SREBP1a, SREBP1c, and SREBP2, are involved in a myriad of physiological and pathophysiological processes ranging from canonical functions, such as transcriptional regulation of fatty acid biosynthesis, endoplasmic reticulum (ER) stress formation, apoptosis, and autophagy, highlighting their distinctive role in lipid synthesis and metabolism
[5]. SREBP cleavage activating protein (SCAP), an ER sensing protein, plays an inevitable role in the regulation of SREBP activity by transporting SREBP from the ER to the Golgi apparatus, facilitating the release of the
N-terminus of SREBP by site-1 and site-2 proteases and permitting its entry into the nucleus. In this compartment, the
N-terminal domain activates the transcription of several genes involved in lipid synthesis, such as HMG-CoA reductase, HMG-CoA synthase, fatty acid synthase, and glycerol-3-phosphateacyltransferase, when cellular cholesterol levels are low
[6]. On the other hand, when cholesterol is present in large quantities in the ER membrane and exceeds the sharp threshold of 4–5% of total lipids, it binds to SCAP, thereby triggering a conformational change promoting SCAP interaction and formation of a ternary complex with the ER membrane retention protein insulin-induced gene 1 (INSIG1), which is composed of 277 amino acids, or insulin-induced gene 2 (INSIG2), which is composed of 225 amino acids. This reduces the transport of the SCAP/SREBP complex to the Golgi, thereby inhibiting the synthesis of cholesterol and initiating a negative feedback mechanism that maintains cholesterol homeostasis
[7].
2. Structure of SCAP
SCAP functions as an ER-cholesterol-sensing membrane protein. It is composed of 1276 amino acids, with a molecular mass of 140 kDa, and is divided into two functional regions
[8]. The first functional region includes the
N-terminal transmembrane (TM) region, consisting of 735 amino acids and eight defined TM helices separated by hydrophilic loops. Loop 1 has cholesterol-sensing properties, seven of which are oriented towards the ER lumen while all other TM helices are faced toward the cytosol. Loop 6 contains a hexapeptide sequence (MELADL) that has an affinity for the coat protein complex II (COPII), which is relevant in the clustering of ER proteins into coated vesicles for transport to the Golgi apparatus
[9]. The second functional region is the soluble C-terminal domain, consisting of 540 amino acids, extending into the cytosol, and including four distinct WD40 protein-interacting repeats, mediating the binding to SREBPs and enabling the formation of a stable SCAP/SREBP complex
[10].
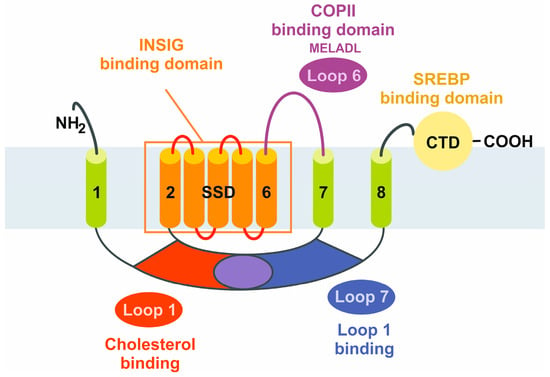
In short, SCAP can be classified as a protein with five functional subdomains: (i) loop 1, with the cholesterol-binding domain and the TM helices 2–6 that form the sterol sensing domain and contain the (ii) INSIG-binding domain, sharing homology with other cholesterol regulatory proteins (iii) the COPII-binding hexapeptide, recognized by a wide array of cargo proteins, such as SEC24, (iv) loop 7, that has affinity for loop 1, and (v) the carboxy-terminal domain (CTD) containing the four WD40 repeats mediating SREBP binding (
Figure 1)
[7].
Figure 1. Schematic structure of the sterol element binding protein cleavage activating protein (SCAP). The SCAP protein is composed of five functional subdomains that are relevant for the binding of cholesterol, insulin-induced genes (INSIGs), coat protein complex II (COPII), and SREBPs, or for sensitizing cholesterol concentrations. The sterol-sensing domain (SSD) mediates the responsiveness towards cholesterol. The eight transmembrane domains are numbered from the N-terminus to the C-terminus. CTD, carboxy-terminal domain.
To elaborate further, SCAP has eight TM helices and INSIG has six TM helices, respectively. The TM helices of SCAP on the cytosolic side are connected by short loops, except for loop 6 which is located between TM 6 and TM 7, and has approximately 94 residues
[11]. All luminal loops in SCAP, except loop 1 located between TM 1 and TM 2 (~239 residues) and loop 7 between TM 7 and TM 8 (~176 residues), are short. On the other hand, all TM helices in INSIG are connected by short loops, suggesting that these TM helices dominate the contact between SCAP and INSIG
[11].
The formation of dimers between SCAP and INSIG is the key control element in the regulation of cholesterol balance in mammalian cells
[11]. When cholesterol levels in the cell exceed a certain threshold the SREBP2/SCAP complex is anchored to the ER through a sterol-dependent interaction between the sterol-sensing domain of SCAP and INSIG. This interaction is more sensitive to hydroxylated cholesterol derivatives, such as 25-hydroxycholesterol (25HC). Consequently, the structure of the SCAP/INSIG/25HC ternary complex is of major importance for the development of novel therapeutics. This ternary complex interacts with Sec23/24, which are basic functional units of COPII coat proteins, in a secretion-associated RAS-related GTPase 1 (SAR1)-dependent manner through an ER export signal located in the third cytoplasmic loop. The binding of the Sec23/24/SRA1 complex is prohibited when INSIG binds SCAP, subsequently preventing SCAP/SREBP binding
[12]. The 25HC molecule is sandwiched between SCAP and INSIG2 in the luminal leaflet of the membrane and the unwinding of a central segment of SCAP in the middle of the membrane is a crucial step required for the formation of the 25HC binding pocket
[13]. The preference of 25HC over cholesterol in establishing SCAP/INSIG interaction is explained by the fact that the binding site is mainly constituted by hydrophobic residues in TM3 and TM4 of INSIG2 and segment 4 to segment 6 in SCAP
[13]. The 25OH group at the end of the 25HC tail is exposed to the cytosolic milieu through a hydrophobic cavity enclosed by SCAP and INSIG2. The limited contact between segment 5 of SCAP and INSIG TM4 causes a fenestration on one side, resulting in a cavity that exposes the 25-OH group of 25HC to the cytosol. The end of the iso-octanol tail creates a hydrophilic environment, explaining the preference of 25HC over cholesterol in facilitating SCAP/INSIG2 interaction.
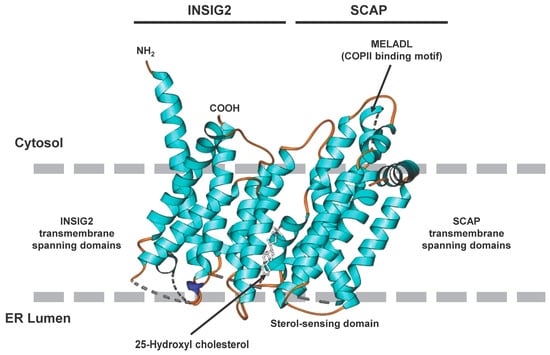
The structure of SCAP/INSIG2 that is formed in the presence of sterols, such as 25HC, solved by cryogenic electron microscopy is depicted in Figure 2, illustrating the interaction of the TM regions and the locations of the sterol-sensing domain orientated to the ER lumen and the location of the COPII binding domain orientated to the cytosol.
Figure 2. Cryogenic electron microscopic structure of the transmembrane region of the human SCAP and INSIG2 complex formed in the presence of 25-hydroxycholesterol. The sterol is sandwiched in a hydrophobic central pocket formed between SCAP and INSIG2 in the luminal leaflet of the membrane. The transmembrane spanning domains of INSIG2 and SCAP anchor the complex in the endoplasmic reticulum membrane, while the COPII binding site, which includes the MELADL motif, is exposed to the cytosol. More details about the complex resolved at resolutions of 3.3 to 3.9 Å are given elsewhere
[13]. The image was generated with Ribbons XP Version 3.0 using coordinates deposited in the RCSB Protein Data Base (access. no.: 6M49)
3. Structure of SREBPs
Out of the three SREBPs that regulate the synthesis of fatty acids and cholesterol, SREBP1c and SREBP1a are translated from a single gene on human chromosome 17p11.2 through the usage of alternate transcription start sites producing alternate forms of exon 1, whereas SREBP2 is derived from a separate gene located on human chromosome 22q13
[14][15][16][17].
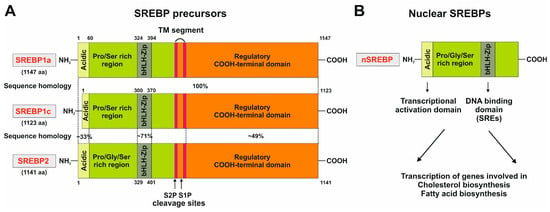
The SREBPs are synthesized as long precursors of 1123–1147 amino acids in length. The
N-terminal part (~480 amino acids) contains a basic helix-loop-helix-leucine zipper. The middle segment, composed of 80 amino acids, has two membrane-spanning domains separated by a short hydrophilic sequence of 30 amino acids. The C-terminal segment consists of 590 amino acids (
Figure 3A)
[14][18].
As the first step to initiate the transcription of genes involved in cholesterol and fatty acid biosynthesis a protease recognition site in SREBP (site 1), which is in the middle of the luminal loop in SREBP, is cleaved by a protease, leading to the breakdown of covalent bonds between two membrane-spanning domains, followed by a second protease cleavage at site 2, which is located in the middle of the first membrane-spanning segment. Consequently, this leads to the release of the NH
2 terminal segment, permitting its entry into the nucleus and allowing binding of the SREs of promoters encoding enzymes involved in cholesterol biosynthesis and LDL receptors (
Figure 3B)
[19].
Figure 3. Domain structure of the three SREBP family members SREBP1a, SREBP1c, and SREBP2. (
A) The precursors of the three members of the SREBP family are comprised of three domains, namely, an NH
2-terminal domain composed of an acidic domain, a proline and serine-rich stretch, and a basic helix-loop-helix-leucine zipper domain, two hydrophobic transmembrane segments separated by a short loop of 30 amino acids, and a carboxy-terminal regulatory region. This part of the figure was redrawn in modified and extended form from
[20]. (
B) The processed nuclear SREBP (nSREBP) contains only the
N-terminal part of the precursor, composed of the acidic domain acting as a transcriptional activation domain and the basic helix-loop-helix-leucine zipper motif that binds sterol response elements (SREs) and acts as a DNA-binding domain. For more details see text.
A cluster of putative binding sites for several transcription factors harboring a nuclear factor Y (NF-Y) site, an E-box sequence, an SRE and an SP1 binding site is at −90 bp of the SREBP1c promoter
[21][22]. In particular, it was suggested that the NF-Y site and the SRE are majorly responsible for SREBP activation
[21]. The sequence motifs in the murine SREBP1 promoter include SRE (5′-TCACNCCAC-3′) and E-box (5′–CANNTG-3′)
[21][23]. The domain structure of the three members of the SREBP family, and the steps involved in the proteolytic activation of SREBPs, are illustrated in
Figure 4.
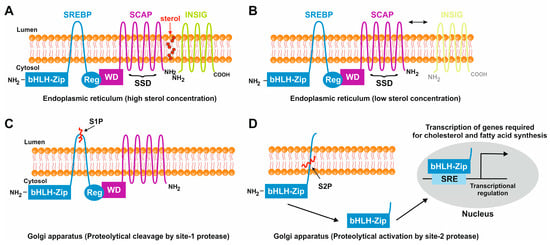
Figure 4. Sterol-mediated proteolytic activation of SREBPs. (
A) In conditions with abundant sterol content, SREBP, SCAP, and INSIG are anchored in the membranes of the endoplasmic reticulum. Under these conditions, SREBP has a hairpin orientation in which both the amino-terminal basic helix loop helix transcription factor motif and the carboxy-terminal regulatory domain are faced to the cytoplasm, while SCAP and INSIG bind to each other and form a hydrophobic cavity for sterol. (
B) In conditions in which the sterol concentrations are low, the binding of SCAP and INSIG gets lost, and SCAP undergoes a conformational change that exposes a MELADL motif which is recognized by COPII. This protein serves as an escort protein, moving SCAP and bound SREBPs to the surface of the Golgi apparatus. (
C) There, SREBP is proteolytically cleaved by the site-1 protease in the 30 amino acid stretch that links the transmembrane domains. (
D) The site-2 protease cleaves SREBP within the membrane-spanning helix. This leads to the release of the carboxyl-terminal part of SREBP which contains the bHLH-Zip motif that shuttles to the nucleus where it activates the transcription of genes that are necessary for cholesterol and fatty acid biosynthesis.
4. Regulation of De Novo Lipogenesis by SCAP/SREBP1c
Under normal physiological conditions, the SREBPs are central and ubiquitous modulators of lipid homeostasis
[24]. The controlled transport of SREBP from the ER to the Golgi is an essential step in the regulation of cholesterol synthesis and other membrane lipids in mammalian cells. Therefore, SCAP has a crucial dual role as a sterol-sensing protein and as a cleavage-activating protein that is required for the transport of SREBPs from the ER to the Golgi apparatus. In sterol-depleted cells, the SCAP-mediated transport of SREBP to the Golgi apparatus is followed by the release of an amino-terminal domain of SREBP that enters into the nucleus and activates lipogenic target genes
[25].
Moreover, the transcriptional regulation of SREBP1c and SREBP2 involves a feed-forward loop in which a feed-forward regulation is mediated by sterol-responsive elements present in the promoters of each gene, promoting their own expression
[22][26].
The selective regulation of SREBP1c, which is the abundant isoform in liver and adipose tissue, occurs by three factors, namely, liver X receptor-α (LXRα), LXRβ, and nuclear receptors, forming heterodimers with retinoid X receptors that are activated by various sterols
[27][28][29]. The SREBP1c promoter has an LXR binding site that activates the transcription of SREBP1c in the presence of LXR agonists, indicating that LXR plays a key role in the insulin-dependent stimulation of SREBP1c
[28]. The enzymes catalyzing the hepatic fatty acid synthesis, acetyl-CoA-carboxylase (ACC) and fatty acid synthase (FAS), are controlled at the transcriptional level via SREBP1c activation by insulin and via carbohydrate-responsive element-binding protein (ChREBP) that is activated by glucose metabolites.
From the three SREBP isoforms, SREBP1a is the most potent activator of all SREBP-responsive genes involved in cholesterol, fatty acid, and triglyceride synthesis, while SREBP1c has a more selective role in enhancing fatty acid synthesis and not cholesterol synthesis. SREBP2 preferentially activates genes involved in cholesterol synthesis, such as HMG-CoA reductase, HMG-CoA synthase, and squalene synthase, that catalyze the biosynthesis of the cholesterol precursor squalene
[30].
Moreover, there is growing evidence that the concentration of several substances, such as sphingomyelin or polyunsaturated fatty acids (PUFAs), affects the protein processing of SREBPs. For example, it has been shown that the depletion of sphingomyelin in CHO cells blocks the proteolysis of SREBPs
[31]. Conversely, PUFAs inhibit the proteolytic processing of SREBPs in murine livers
[32], ultimately reducing cholesterol synthesis and uptake (
Figure 5).
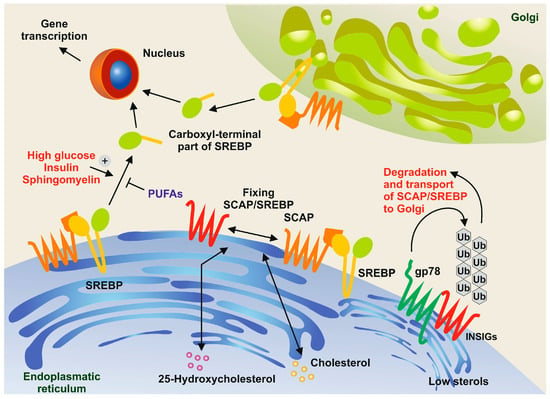
Figure 5. Sphingomyelin and polyunsaturated fatty acids (PUFAs) in the control of SREBP processing. In conditions with high concentrations of cholesterol and 25-hydrocholesterol, the sterol regulatory element-binding protein (SREBP) and the SREBP cleavage-activating protein (SCAP) form a complex and are retained in the endoplasmic reticulum along with insulin-induced gene proteins (INSIGs). However, when sterol concentrations are low, membrane-bound ubiquitin ligases, such as gp78, bind to INSIGs, leading to their rapid degradation through the ubiquitin pathway
[33]. In turn, SREBPs are transported to the Golgi apparatus. The N-terminal domains of SREBPs are then released through the action of site-1 protease (S1P) and site-2 protease (S2P). These domains move to the nucleus, where they activate the expression of genes necessary for cholesterol and fatty acid synthesis. This proteolytic processing is triggered by high concentrations of glucose, insulin, and sphingomyelin, while it is inhibited by PUFAs.
5. The Central Role of SCAP/SREBP1c in MASLD Pathogenesis
The widely accepted ‘multi-hit’ theory of MASLD pathogenesis was derived from early studies suggesting excessive fatty acids contributing to hepatic steatosis and insulin resistance as the first hit and subsequent damage of hepatocytes due to inflammation, fibrosis, and other pathological changes due to oxidative stress, lipid peroxidation, ER stress, and lipotoxicity as the second hit
[34]. In this scenario, it is obvious that the imbalance between lipid synthesis, availability, and lipid disposal by fatty acid oxidation and VLDL secretion is the fundamental cause of steatosis
[35].
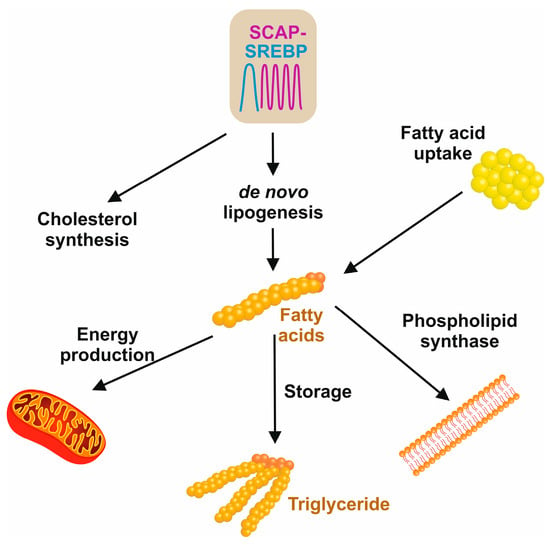
The three main sources of fatty acids in the liver include hepatic de novo lipogenesis, free fatty acids from adipose tissue, and the uptake of dietary fats (
Figure 6)
[36]. The hallmark feature of insulin resistance and obesity states in MASLD is that the content of hepatic fatty acids derived from de novo lipogenesis contributes about 26%, in contrast to 5% in normal healthy individuals
[36].
Figure 6. Sources and cellular usage of fatty acids. De novo lipogenesis and cholesterol synthesis are driven by the SCAP/SREBP axis. The pool of fatty acids is further increased by dietary free fatty acids that are released by the hydrolysis of triglycerides. In the cell, fatty acid is used for the synthesis of phospholipids in membrane biogenesis, mitochondrial fatty acid β-oxidation to maintain cellular energy metabolisms, or the synthesis of triglycerides that, in excess of fatty acids, can be stored in the liver or in fat cells to supply the body with energy when it is required.
SREBP1c is a critical modulator involved in the regulation of hepatic de novo lipogenesis by insulin by preferentially activating genes involved in fatty acid synthesis, such as fatty acid synthase, elongation of very long chain fatty acids-like 6 (ELOVL6), ATP-citrate lyase, acetyl CoA carboxylase, malic enzyme, glucose 6 phosphate dehydrogenase, stearoyl-CoA-desaturase, and glycerol-3-phosphate acyl transferase, whereas SREBP2 preferentially activates genes required for cholesterol synthesis
[37].
The cleavage and transcriptional activity of SREBP are inhibited by AMP-activated protein kinase (AMPK). This conserved enzyme is an energy sensor playing a key role in cellular energy homeostasis and its stimulation suppresses SREBP cleavage and nuclear translocation attenuating hepatic steatosis, as demonstrated in LDL-deficient mice with diet-induced insulin resistance
[38].
Similarly, ER stress promotes lipogenesis by activating SREBP1c, leading to the progression of MASLD and severe forms of MASH. The unfolded protein response (UPR) is linked to inflammation and hepatocyte apoptosis in MASH, and this claim is supported by the finding that hepatic overexpression of the heat-shock protein-70 family member glucose regulated protein 78 (GRP78), which is critically implicated in the folding and assembly of proteins in the ER, reduced markers of ER stress in ob/ob mice by inhibiting the cleavage of SREBP1c and SREBP2 target genes
[39].
Another factor relating SREBP to the pathogenesis of MASLD is lipotoxicity in hepatocytes, which provokes hepatocyte damage when lipotoxic substances are elevated beyond the hepatocyte’s ability to transport them.
Liver fibrosis is characterized by the excessive deposition of extracellular matrix components between hepatocytes and sinusoids, causing liver stiffness and distortion of the liver architecture. Activated hepatic stellate cells in the liver produce collagen, which is an important component of the extracellular matrix network. TGF-β activates signaling pathways, such as MAPK, mTOR, PI3K/AKT, and Rho/GTPase, leading to liver fibrosis. These pathways are regulated by SREBP1c, which impacts hepatic stellate cell activation
[40].
Strikingly, SREBP2 has also been implicated in liver fibrosis through the regulation of cholesterol levels in hepatic stellate cells. For example, in mouse models of hepatic steatosis and in primary mouse hepatic stellate cells, the nuclear form of SREBP2 increases with hepatic stellate cell activation
[41].
6. Conclusions
De novo lipogenesis, the metabolic pathway that synthesizes saturated fatty acids and monounsaturated fatty acids from acetyl-CoA, only accounts for a small fraction of fatty acids in the liver in a healthy human population. In lean individuals, this fraction is typically around 5%. However, in individuals with MAFLD, the rate of de novo lipogenesis is significantly increased, with 25% of triglycerides arising from de novo lipogenesis. The expression of de novo lipogenesis enzymes in the liver is influenced by dietary lipids and nutritional state. However, the lipogenic transcription factors, and, consequently, DNL, are constantly active. The chronic energy excess caused by both glucose flux and hyperglycemia acts as a driving force in elevating triglyceride synthesis from saturated fatty acids and monounsaturated fatty acids. Therefore, the induction of de novo lipogenesis through SREBP1 suggests a strong link between de novo lipogenesis, SREBP1, and the progression of MAFLD
[42].
MASLD progression to MASH has a multitude of detrimental effects and is one of the leading causes of liver disease worldwide affecting one-third of the world’s population. Multiple studies have documented the central role of the SCAP/SREBP axis in regulating lipid homeostasis. Thus, targeting the SCAP/SREBP axis may be an attractive and powerful option in treating MASLD. SCAP, a cholesterol sensor, impacts MASLD and other metabolic diseases. It is an indispensable protein required for the transport and activation of all three SREBP isoforms. SCAP and SREBPs are intricately linked to numerous signaling pathways, contributing immensely to the pathogenesis of MASLD. Therefore, pharmacological modulation of SCAP/SREBP activation by small-molecule inhibitors can be an attractive option for treating this complex disease with its wide spectrum of clinic-pathological conditions.
This entry is adapted from the peer-reviewed paper 10.3390/ijms25021109