Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Sickle cell disease (SCD), is a congenital blood disorder characterized by considerable phenotypic diversity. It comprises a group of disorders, with sickle cell anemia (SCA) being the most prevalent and serious genotype.
- sickle cell
- hemoglobin
- anemia
- microbiota
- nutrition
- vaso-occlusive crisis
1. Introduction
Sickle cell disease, an often overlooked disease in the 21st century, is a noncontagious and enduring congenital blood disorder. It encompasses a group of clinical syndromes that affect hemoglobin due to a genetic code for abnormal polymerized deoxygenated hemoglobin. This abnormal hemoglobin distorts the shape of red blood cells, and it is inherited by children from their parents [1]. The term sickle cell disease (SCD) is derived from the polymerization of two mutant sickle β-globin subunits leading to a crescent or sickled shape of erythrocytes [2].
Sickle cell disease comprises various genotypes, yielding a group of hemoglobinopathies [3]. The production of hemoglobin is regulated by the inheritance of a pair of genes, but there is considerable variability in absolute hemoglobin levels among patients with SCD [4]. Sickle cell anemia results from the inheritance of two sickle genes, with one gene from each parent [5][6].
Two parts, heme and globin, constitute the normal form of hemoglobin. The protein is made up of four polypeptide chains (two α chains and two β chains). There are many known mutations in the hemoglobin subunit β-HBB (β-globin protein) coding gene, which make up the most common form of hemoglobin in adult humans, hemoglobin A (HbA) [7]. A variety of inherited diseases arise from these mutations. Abnormal versions of β-globin, such as hemoglobin C (HbC), hemoglobin E (HbE), and hemoglobin S (HbS), are produced by a variant mutation in the HBB gene. It is this mutation in the HBB gene that causes sickle cell anemia [8].
Sickle cell anemia (SCA) is the most prevalent and serious genotype of SCD, followed by HbSC (“mild” form of SCA), hemoglobin (Hb) Sβ thalassemia, HbSβ+thalassemia (accounting for some 30–40% of SCD patients), and other rare and benign genotypes [9][10].
Sickled red blood cells are susceptible to chronic hemolysis [11], and emerging evidence reveals that SCD is made evident by the presence of chronic inflammation and oxidative stress, both of which play a role in the development of chronic vasculopathy and several other enduring complications [12]. SCA, characterized by abnormal red blood cells and hemoglobin, is worsened by low oxygen levels in the air [13].
SCA is manifested as the result of the presence of an autosomal recessive allele, which is found on the short arm of chromosome 11p15.5 [14]. This alteration of the genetic code leads to the substitution of a single amino acid, where valine replaces glutamic amino acid in the sixth position of the 146 amino acids of the β chain of hemoglobin [5] (Figure 1).
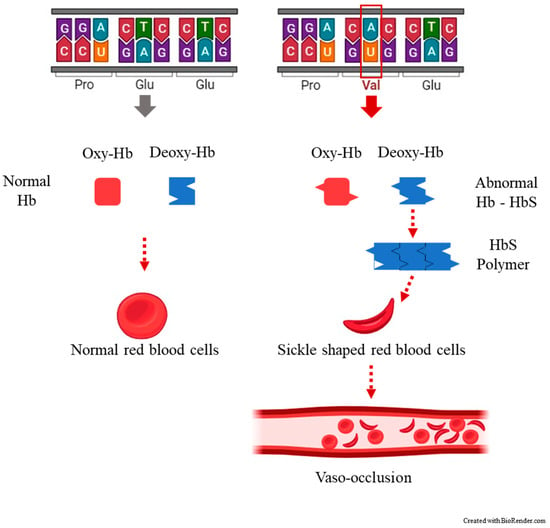
Figure 1. The most common clinical manifestation of sickle cell disease, a vaso-occlusive crisis (VOC) occurring when blood flow is blocked by sickled red blood cells (crescent-shaped) to the point that tissues and organs become deprived of oxygen, causing pain.
After more than one hundred years since the discovery of sickle cell group of hemoglobinopathies as genetically inherited diseases [15], new studies are still necessary to explore the molecular mechanisms leading to fetal hemoglobin induction and find ways to reduce the adverse effects in patients with SCA and other β-hemoglobinopathies [16].
There are both severe and mild SCD genotypes, which reflect the type of symptoms and prognoses for the disease. Research has been conducted during routine patient care to identify possible clinical biomarkers among SCD patients. These biomarkers may vary according to genotype and treatment categories. However, there is still insufficient progress in developing treatment options or counseling decisions [17][18][19].
Sickle-shaped red blood cells are more rigid and stickier, which leads to the obstruction of small blood vessels. This obstruction prevents oxygen from reaching body tissues and organs, inducing both acute and chronic intense pain. There is little research focusing on the pathophysiology of acute or chronic pain in SCD, and therefore it is still poorly understood. However, it is believed to be dependent on the interaction of several molecular mechanisms [2][20][21].
1.1. The Incidence of Sickle Cell Disease
This disease substantially induces multimorbidity and impairs quality of life, while placing strain on healthcare systems wherever it exists [22][23]. The global burden of this disease has been assessed [24], highlighting the high risk of child mortality associated with SCA. In Sub-Saharan Africa, it can contribute to as much as 90% of under-5 mortality [25][26], with approximately 500 children with SCD continuing to die prematurely every day [27]. This is due to delayed diagnosis and/or the lack of access to comprehensive care, a trend that urgently needs to be reversed [9].
Every year, between 300,000 and 400,000 newborns with SCA are delivered around the world, whereas tens of thousands of people show the homozygosity for hemoglobin S form, which represents the most severe clinical phenotype of the disease [28]. Although SCD occurs worldwide, Sub-Saharan Africa is the region with the highest prevalence. It is estimated that approximately 1000 children with SCD are born in Africa every day, and more than 500 of them die before reaching the age of 5 years [29].
Children suffer several preventable chronic disorders that are followed by premature death associated with SCD. Efforts have been made to identify achievable goals to improve outcomes both in the short and long term. These initiatives aim to recitfy the present unfair attention given to this inherited condition, particularly in developing countries [30].
Approximately 1 in 12 African Americans carries the SCD mutation, and 1 in 500 African Americans suffers from the disorder. In the U.S., 1 out of every 16,300 Hispanic-American neonates is born with SCA each year [31]. Epidemiological data on all blood disorders is still scarce, but SCD is estimated to affect approximately 250 million people globally.
Despite the increase in the global burden of SCD, which is believed to affect over 20 million people [32], including an estimated 200,000 annual sickle genotype births in Sub-Saharan Africa [33], available data on SCA prevalence, morbidity, and mortality remains limited on a global scale. However, some systematic reviews on global data exist [34][35].
Different areas in Côte d’Ivoire, Egypt, Lake Chad, Sudan, Lake Victoria, the coast of Kenya, Tanzania, Mozambique, and the east coast of Madagascar have been projected to have a a predicted HbS allele frequency between 7.5% and 12.5% [36]. In northern Mozambique, hematological studies have revealed a prevalence of sickle cell trait (HbAS) and G6PD (glucose-6-phosphate dehydrogenase) deficiency to be around 4% [37][38].
Sickle cell trait (HbAS) is notably more common in West Africa. It is very interesting and well-known that carriers of the sickle cell trait HbAS experience natural and nearly complete protection against severe Plasmodium falciparum malaria. This protection is observed despite the inadequately understood relationships between HbAS, malaria, and other common causes of child mortality [39][40][41][42].
1.2. Sickle Cell Disease Physiopathology
When cells are subjected to physiological stressors, they react with a mechanism described as the heat shock response. This mechanism activates a certain type of critical molecular regulator called heat shock proteins (HSPs) [43].
Heme oxygenase 1 is a member of the heat shock protein (HSP32) family and is involved in numerous cellular operations [44]. Increased heme in SCD causes the upregulation of heme oxygenase 1, which leads to cardiomyopathy through ferroptosis, an iron-dependent nonapoptotic form of cell death [45].
This enhances the fact that both genetic and environmental factors affect the process. Thus, the understanding of biomarkers and the molecular basis of diseases such as SCA are significant in playing a definitive role on the onset of such pathologies and, therefore, on the prevention strategies [46][47].
This leads to the formation of hemoglobin S and the change to sickle-shaped red blood cells compared to normal red blood cells. These cells obstruct the bloodstream, hence leading to serious problems, including cerebrovascular accident, nephropathy, retinopathy, infections, aches, and pains [48] (Figure 2).
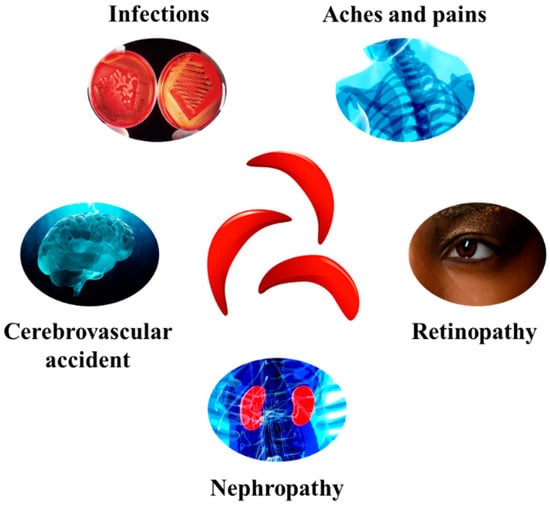
Figure 2. Some of the major complications associated with SCD development.
Among the four DNA bases (two purines: adenine and guanine, two pyrimidines: thymine and cytosine), guanine has the lowest redox potential and is preferentially targeted for oxidation [49]. Despite guanine reduced redox potential, guanine radicals are known to trigger mutations and damage the genetic code, which are involved in carcinogenesis and ageing [50].
Because SCD is hallmarked by an underlying chronic inflammatory status, which is partly driven by proinflammatory M1 macrophages [51], heme scavenging or modulation, as well as the potential therapeutic targeting of mitochondrial biogenesis, might significantly ameliorate tissue damage associated with SCD pathophysiology [52].
Peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC1α), a transcriptional coactivator protein that regulates the genes involved in energy metabolism [53], exerts significant control over, induces, and coordinates gene expression. It stimulates mitochondrial oxidative metabolism (i.e. respiratory capacity, oxidative phosphorylation, and fatty acid β-oxidation), produces ATP and lipids, induces amino acid and heme biosynthesis, and generates/sequesters reactive oxygen species (ROS) [54].
1.3. Sickle Cell Disease Diagnosis
Sickle cell disease can be prevented prenatally, and it can also be diagnosed in utero or in the newborn period through screening. Early diagnosis of this condition is essential for beginning treatments that can reduce the risk of life-threatening complications, such as severe infections and strokes, as well as managing the disease effectively to reduce morbidity. SCD is diagnosed through a simple complete blood test, peripheral blood smears, hemoglobin electrophoresis, HPLC, and various genetic sickling tests. Hemoglobin S solubility assay and sodium metabisulfite test may be used for screening individuals aged 6 months or older. For pregnant women, screening should ideally be conducted before 10 weeks’ gestation. Recent studies have also reviewed current emerging portable techniques that have been developed for the early detection and diagnosis of sickle cell disease and carrier states [55][56][57]. More detailed molecular genetic diagnose testing is also available [58].
2. Current Treatments of Sickle Cell Disease
Increasing the production of fetal hemoglobin (HbF) in significant quantities can diminish the severity of the clinical progression in β-thalassemia and SCD. This can lead to a decrease in morbidity, disability, impairment, illness, and mortality [59].
There are science-based guidelines elaborated by the American Society of Hematology (ASH) designed to support patients, clinicians, and other healthcare professionals, namely, in pain management decisions for children and adults with SCA. However, these do not provide specific guidance on nutritional care and other strategies [60][61].
SCD is caused by a mutation that results in the substitution of glutamic acid for valine.
Until recently, only the use of an oral chemotherapeutic drug, hydroxycarbamide (also known as hydroxyurea), was considered for the treatment of SCD. Hydroxyurea, which is a ribonucleotide reductase, is the only approved drug for disease-modifying treatment in patients with SCA [62]. However, it is currently underutilized in clinical practice [63].
A class of new medications called hemoglobin S (HbS) polymerization inhibitors (e.g., voxelotor), has been recently approved by the FDA in 2019 and by the E.U. EMA in 2022. These drugs are intended for the oral treatment of hemolytic anemia due to SCD and vaso-occlusive crisis (VOC), in adults and children aged 12 years and older [64].
This small-molecule drug is able to attach to and stabilize hemoglobin, preventing hemoglobin polymerization (i.e., formation of abnormal hemoglobin) that causes the formation of sickle shaped red blood cells [65]. In well-resourced countries, three potential treatments are available for preventing or reducing the morbidity and mortality associated with SCA: transfusions, hydroxyurea, and stem cell transplantation [66]. There is no evidence of any benefits of corticosteroid use in SCD acute events [67].
The polymerization of abnormal hemoglobin S upon deoxygenation in the tissues to form fibers in red cells causes the development of SCD, thus, generating deformations and blockages in the circulation. Hence, many attempts have been made to find drugs that can control nonpolymerizing fetal hemoglobin [65].
Vaso-occlusive crisis has been prevented and treated using an approved drug called crizanlizumab. This drug is designed to treat pain by preventing blood cells from sticking to the inner walls of blood vessels. The monthly administration of this monoclonal antibody against P-selectin (mediator of inflammation through promoting adherence of leukocytes to activated platelets and endothelium) has proven effective in lowering the frequency of sickle pain crises [68].
There is a hypothesis, which requires further investigation, suggesting that leucine transcriptional nuclear factor NRF2 activation with sulforaphane (a chemical compound found in vegetables such as broccoli and Brussels sprouts), may offer therapeutic benefits for SCD patients. These potential benefits could include reducing liver damage, restoring oxidative capacity, and increasing fetal hemoglobin concentration [69].
Allogeneic hematopoietic stem cell transplantation, also referred to as bone marrow transplant, has been known to cure severe congenital anemias. This treatment has been used to transplant healthy hematopoietic stem cells, obtained from several sources, into patients with dysfunctions related to many malignant and nonmalignant disorders [70] (Table 1).
Table 1. Therapeutic interventions in sickle cell disease.
| Therapeutic Intervention | Outcome |
|---|---|
| Blood transfusion | Reduce the burden of sickled cells [71][72] |
| Hydroxyurea | Increase fetal hemoglobin (HbF) to stop polymers forming in the sickle hemoglobin [73] |
| Hematopoietic stem cell transplantation | Reverse the sickle phenotype [74][75] |
| L-glutamine | Antioxidant effects [76] |
| Hemoglobin S (HbS) polymerization inhibitors | Prevent HbS polymerization [77] |
| Monoclonal antibody (crizanlizumab) | Reduce selectin-mediated adhesion [78] |
| Gene editing therapy (Casgevy™) | Editing faulty gene in a patient’s bone marrow stem cells [79] |
Recent Advanced Therapeutic Approaches for Sickle Cell Disease
Currently, the sole validated healing medicinal approach for SCD is allogeneic (genetically dissimilar) hematopoietic stem cell transplantation, ideally from an unaffected human leucocyte antigen (HLA)-identical matching sibling donor, when available. Combining genetic treatments and hydroxyurea seems to provide the best results [80]. However, normally, this is only considered for children younger than 16 who have severe complications from the disease [81][82][83][84].
Although still under review in the U.S., the European Union, and Saudi Arabia, the Medicines and Healthcare products Regulatory Agency (MHRA) in the U.K. has conditionally approved an innovative and first-of-its-kind gene editing therapy for patients aged 12 and over with SCD and transfusion-dependent beta thalassemia. It is worth noting that in the U.K., there are approximately 15,000 SCD patients.
These treatments, known as CasgevyTM and LyfgeniaTM, which have been approved by the FDA, are designed to work by editing the faulty gene in a patient’s bone marrow stem cells. They are the first medicines to be licensed using the innovative gene editing tool CRISPR. The inventors of LyfgeniaTM were awarded the Nobel Prize in 2020 [79]. Following the U.K.’s authorization to use CasgevyTM, Bahrain was the second country to approve the use of the drug, and in December 2023, the U.S. became the third.
A drug called masitinib, which has been available in Europe since 2008 for veterinary use under the name “masivet”, a tyrosine-kinase inhibitor used in canine cancer treatment, is presently being tested for the treatment of SCD. A new clinical development program for a phase two clinical trial of masitinib in SCD has been granted a substantial investment to study the involvement of mast cells and basophils in orchestrating acute and chronic complications of SCD. A new patent has been filed, which, if granted, will extend the international protection of masitinib for use in treating SCD until 2040 [85].
Microglia from the brain and spinal cord and mast cells (bone marrow-derived tissue-dwelling cells), two key immune cells involved in numerous pathologies, release substances such as histamine, leukotrienes, and cytokines, causing inflammation. This process is strongly related to SCD [86].
3. Influence of Gut Microbiome in Sickle Cell Disease
The gut microbiota, their genes (microbiome), and metabolites express a large number of potential signaling molecule ligands and metabolites. These components, under homeostatic conditions, can control inflammatory interleukins, growth factors, cytokines, and prostaglandins in coordination with tissue immunity [87][88].
The gut microbiota, which accommodate approximately 1013–1014 cells belonging to a diverse group of microorganisms [89] (equivalent to over 3 million genes), play a key role in human health by determining the development of conditions linked with the nervous system, autoimmunity, metabolism, and inheritance [90][91].
Increased gut injury and permeability, modified microbiota composition, and bacterial overgrowth, along with collateral bacterial translocation in SCA patients, reveals a state of dysbiosis and an increase in gut microbiota responsible for triggering inflammation [92].
Gut microbiota are closely involved in energy homeostasis [93], immune system regulation [94], metabolism [95], and other physiological processes of hosts, as reported by research studies.
A disruption of the microbiome spawns considerable dysbiosis. This dysbiosis is observed in SCD patients because some bacteria trigger proinflammatory responses and can affect some of the pathophysiological features of this disease [96]. This microbial imbalance was mainly recorded in the U.S., as SCD patients often are hospitalized and subject to nosocomial infections. However, in Africa, where SCD prevails, not many studies have been conducted, and the gut microbiome profile is associated with race and/or ethnicity [97].
A complex interplay occurs between environmental (e.g., diet and medications) and host factors within the gastrointestinal tract, where symbiotic microorganisms reside. This interplay is affected by the human genome, which affects the gut microbiome through enzymes and miRNAs [98]. The target for customized nutrition and therapy, as well as the renovation of exchanges between the microbiota and host, and the modification of nutrition to restore the necessary symbiosis, is microbiota modulation. The aim is to reverse established microbial dysbiosis [99][100][101].
A crucial mediator in the immunomodulation of inflammation, cell adhesion, and induction of aged neutrophils, which are the main arbitrators of recurrent VOC, is diverse gut microbiota [102].
Several studies have shown that host genetics affect the composition or structure of the gut microbiome and vice versa [103][104].
To understand the complex pathophysiology of the disease and the evaluation of potential specific therapies, researchers have developed several murine models and generation of transgenic mouse clones as a solution for the absence of a natural animal pattern for SCD [105][106][107]. Most studies on this interaction have therefore been conducted using genetically engineered mouse models.
Therapeutic strategies involving synergistic gene addition and gene silencing in stem cell progeny have demonstrated proof of concept through targeted research [108]. The gut microbiome has been characterized in murine models with SCA, revealing significant dysbiosis [92]. The gut microbiota is believed to play a role in the severity of SCD because the permeability of the intestinal barrier is compromised. This is connected with gene silencing of continuous intercellular network proteins, enhanced inflammation, and oxidative stress, which are all specific to the small intestine [109].
The generation of ROS and other free radicals occurs during normal cellular metabolism, and inflammation and oxidative stress are intertwined, one aggravating the other. The therapeutic potential of natural food antioxidants has been widely researched, namely, in chronic metabolic disorders [110][111].
SCD patients show decreased protection from antioxidants in their blood, possibly due to lipid peroxidation, which results from its interaction with ferroptosis and a compromised antioxidant competence [112]. When administered at appropriate doses, omega-3 fatty acids (ω-3 FAs), specifically docosahexaenoic (DHA) and eicosapentaenoic (EPA) acids, are potent anti-inflammatory mediators that modulate pain. They also decrease episodes of VOC in SCD [113]. The therapeutic effects of polyunsaturated ω-3 fatty acids in SCD have been shown in clinical trials, providing increasing evidence for a safe and effective treatment for SCD, demonstrating improvements in VOC rates, biomarkers of inflammation, cell adhesion, and hemolysis [114][115].
Chronic sickle cell pain and osteoporosis are common clinical manifestations in humans, but their underlying causes are not fully understood. It appears that the gut microbiome may play a role in the management of chronic SCD pain. Increased gut tissue injury and permeability and bacterial translocation of luminal contents contribute to the role of the microbiome in SCD [92][116].
It is also suggested that gut dysbiosis in SCD induces pain through changes in vagal nerve activity [117]. Increased inflammatory cytokines, which are critical mediators that oversee and regulate immune and inflammatory responses via complex networks serving as biomarkers, arise from increased gut bacteria burden and augment antigenic load, travelling across the impaired intestinal barrier through inflammation [118] (Table 2).
Table 2. Nutritional and gut microbiome interventions in sickle cell disease.
The identification of therapeutic approaches for gut modulation is still in its early stages despite the evaluation of the microbial differences between SCD patients and healthy controls in some studies [26][121].
4. Nutritional Perspectives in Sickle Cell Disease
Nutritional imbalances are considered as crucial factors contributing to the severity of sickle cell disease. This has led to increased interest in promoting dietary supplementation for treating patients, especially because no cure for sickle cell anemia is available. Patients with sickle cell disease require higher energy and protein intake compared to healthy individuals. These patients tend to suffer from undernutrition if their energy intake is consistently low [122].
There has been little researched on dietary interventions that may be useful as a supplementary tool to treating SCD. However, it is well-established that unbalanced nutrition is a significant risk factor that adversely impacts clinical events, welfare, vital processes, and the independence of patients [123].
There is a deficiency in knowledge and the possible integration of nutrition into sickle cell medial services. Awareness must be raised regarding the value and importance of the role of nutrition in improving the management and care of SCD, particularly in Africa [124]. Nevertheless, helpful dietary suggestions, especially for children with SCD, must be a priority.
Major factors contributing to the increased severity of SCD include chronic inflammation and oxidant stress. Hence, the establishment of recommended reference intakes for SCA patients is fundamental, and nutritional intervention should be included as supplementary care in conjunction with standard practices [125].
Because no easy and cost-effective treatment has yet been found, recent efforts have been directed toward nutritional interventions to reduce ill health and improve the quality of life for SCD patients. However, there are limitations within the current scope of the recommended daily allowances and dietary reference intakes for world populations, and nutrient density has also been under consideration [126]. This makes it even more challenging to establish specific nutritional requirements for sickle cell patients.
The role of malnutrition as one of the complications of SCA and the possible benefit of regular micronutrient supplements have been recently demonstrated by several authors [127][128]. Symptoms occur around the age of 5 months, but they vary among individuals and are characterized by episodes of pain, fatigue, frequent infections, organ damage, and early mortality. These symptoms can cause delayed growth and development in children while resulting in a need for a higher amount of certain nutrients, including calories and protein [129].
Due to chronic ischemia-reoxygenation damage induced by VOC, patients with SCD may suffer from leaky gut, which affects microbiota density and adhesion to the epithelial wall and the degree of translocation [130]. This influences nutrient intake, metabolic homeostasis, hormonal environment, microbiota imbalance, and immunity [131].
Particular attention has been directed to identifying dietary deficiencies that often coexist with SCD and searching for novel dietary strategies to reduce morbidity and improve the quality of life for these patients [132][133]. SCD has been also associated with vitamin D deficiency and poor appetite. Although a few studies have been conducted, none have been of sufficient quality to guide clinical practice [134].
Recognizing gene–nutrient interactions with the objective of explaining a specific response in various ethnic and environmental contexts is a complex task [135]. The prevalence of nutritional deficiencies in SCD is evident, and they may be associated with worse pain outcomes [96]. Nutrition, being a complex and wide-ranging issue, calls for the implementation of a new collaborative approach that addresses staple food types and sources, micronutrients, and phytonutrients, which are all critical for optimal human health and well-being. Particular focus should be given to improved digestibility, the human gut microbiome, overall vitality, and mental health [136][137].
Children with hereditary diseases such as SCD frequently have feeding disorders and dysphagia as a result of the elaborate interplay between anatomical, physiological, medical, and behavioral factors. These feeding complications may also cause food consumption to be troublesome, passive, or even painful because of a lack of breath and ability to talk, choking, coughing, tiredness, or vomiting, causing the child to stop eating and requiring a parent to feed their child.
Dietary intake has been inadequately considered in SCA, and questionable guidance for dietary iron restriction has been assumed, although iron deficiency is unexpected in SCD patients based on the fact that the homozygous SCA genotype is associated with the most severe form [138].
It has been proven that the disease mechanism of SCD has considerable nutritional and health connotations, including elevated energy and nutrient requirements, nutrient deficiencies, and growth abnormalities [139][140]. However, these data were obtained using small sample sizes, namely in Sub-Saharan Africa, where it prevails. Hence, the need for further investigation of the potential benefits of nutrition-related interventions for these patients is imperative [141]. Over the last decade, the prime concerns for basic, clinical, and demographic research regarding food, diet, supplements, and nutrition in individuals with SCD and thalassemia have been suggested as main areas of research and innovation [142][143].
The main obstacle has been difficulty in the assessment of dietary intake, nutritional status, the use of nutritive and nonnutritive dietary supplements, and increased vulnerability to infections caused by specific pathogens in these patients, particularly children under 5 years of age [144].
The risk of a worse prognosis in SCA has increased due to inadequate food and nutrient intake data in developing countries [145]. Plans to motivate the intake of minimally processed foods should be considered due to the benefits of antioxidants acting positively against SCA [146][147].
Micronutrient deficiencies in these patients may increase vulnerability to stunting, inflammation, opportunistic infection, and acute painful crisis [148]. A number of micronutrient deficiencies and their associations, including iron, zinc, copper, folic acid, pyridoxine, and vitamin E, have been long considered and addressed [149]. While it is possible that folic acid supplementation may increase serum folate levels, the effect of supplementation on SCA remains unclear [150].
Randomized clinical trials have assessed the efficacy of antioxidant nutrient supplementation in reducing hemolysis in SCD patients. It found that vitamin C and E, when used in safe doses, were considered to worsen hemolysis [151]. However, supplementation with ω-3 fatty acids, vitamin A, and zinc were reported to improve indirect hemolytic markers [147].
Some preliminary meta-analyses have found a that supplementation with the semi-essential amino acid L-arginine or its precursors has a beneficial effect on patients with SCD. The conversion of proline, glutamate/glutamine, and the nonproteinogenic amino acid citrulline leads to the endogenous synthesis of L-arginine [152][153], which plays an important role in cell division, the healing of wounds, the stimulation of protein synthesis, immune function, and the release of hormones [154].
Morbid occurrences in SCA lead to an increase in the generation of reactive oxygen species (ROS) through the activation of several prooxidant enzymes. Under normal conditions, these enzymes would be balanced between oxidant and antioxidant systems to prevent oxidative damage. However, the injury of sickle red blood cells causes hemolysis, the release of free hemoglobin, the modification of mitochondrial respiratory chain activity, and red blood cell autooxidation [155].
A surplus of free radicals can damage cells, causing illness and aging and contributing to greater oxidative stress in erythrocytes, endothelial cells between the bloodstream and the surrounding tissues cells, polymorphonuclear leukocytes, and thrombocytes. This oxidative stress is associated with multiorgan disorders, vasculopathy, and cellular dysfunction [156].
Despite the capacity, constraints, and frustrations of antioxidant treatment to date, many foods containing powerful antioxidant enzymes may play an essential role in contributing to the mitigation of oxidative-related obstacles. This enables the development of nutritional strategies aimed at improving antioxidant status in SCA patients [157].
However, free radicals react too quickly with lipids, proteins, and nucleic acids in cell membranes, making it challenging for exogenous monomers to scavenge them effectively [158]. Hence, the idea of collecting these radicals in biological systems using exogenous compounds has been considered impractical [112].
It is not the deficit of essential polyunsaturated fatty acids in the diet, but rather the modified metabolic pathways of fatty acid elongation and desaturation on the endoplasmic reticulum membrane in children with SCD that leads to reduced polyunsaturated fatty acids in the phospholipids of the cell membranes, which contributes to known disease symptoms [159].
The possible nutritional strategy, when used as a complement to adequate treatment, must focus on mediating the mechanism of a free-radical substitution reactions. This involves maintaining a balance between the formation and removal of free radicals, which can be achieved through a diet rich in healthy, high-antioxidant foods [160].
In individuals with SCD, the antioxidant defense system is greatly compromised due to depleted expression and activity levels of antioxidant enzymes (e.g., superoxide dismutase-SOD, catalase-Cat, and glutathione peroxidase-GPx). These enzymes play a crucial role in breaking down hydrogen peroxide and thus controlling its intracellular concentration [161][162].
4.1. African Plant Resources in Sickle Cell Disease
There is the need in Africa to embrace both natural and pharmaceutical medicine, creating a harmony between nature and science and dismissing any stigma associated with sickle cell disease. The exploration of native African herbs and plants for medicinal use has long been the subject of extensive research. In addition, there has been a time-consuming search for new drugs through molecular pharming [163].
Interest in natural products is gaining attention as an integrative approach to the management of sickle cell disease, particularly in Africa, where there is rich biodiversity. As many as 5000 local medicinal foods [164] directly sourced from the wild are used by traditional healers, who serve approximately 80% of the African population [165][166].
Traditional remedies, often based on empirical observations that have proven their accuracy over time, are more popular than pharmaceutical drugs. However, this knowledge is at risk of being lost to younger generations because it is verbally transferred from generation to generation, and they are not eager to inherit this heritage [167]. The bioactive compounds found in these tropical plants interact with the gut microbiota and the available phytonutrients, playing a critical regulatory role in human health [168].
Several structured surveys have been conducted in Sub-Saharan Africa, where approximately 80% of the world’s SCA cases occur. These surveys were designed to assess various challenges and dietary aspects of patients with SCA. Their aim was to identify knowledge gaps and prioritize future areas of research [32][35][141][169].
The potential benefits of nutrition in African SCD patients have been studied primarily in Nigeria [170], with less research in other Sub-Saharan countries, such as Ghana, Sudan, Kenya, Malawi, Tanzania, Cameroon, Ivory Coast, Gabon, and Mali. These studies have explored the use of indigenous medicinal sources, including seed oils from Solenostemon monostachyus, Ipomoea involucrate, and Carica papaya plants, commercial Cajanus cajan plant extracts, and Acacia Senegal seed oil [141] (Figure 3).

Figure 3. Some of the tropical plants used in SCA in Sub-Saharan Africa.
The leaves, root bark, and seeds of Alchornea cordifolia (Christmas bush) and Ceiba pentandra are wild harvested and cultivated for their medicinal uses in DR Congo. They are used to produce a beverage known as “blood tonic” that is used by individual suffering from SCA [171].
Found throughout Africa, Moringa oleifera is rich in many phytochemicals exhibiting antiurolithiatic properties. It is widely used for a vast number of ailments and might be significant in the management of SCD [172] (Table 3).
Table 3. Traditional medicine in sickle cell disease.
A study conducted in Sudan on Nigella sativa (black cumin seed) oil extract, which is known to have calcium antagonist antioxidant properties that are useful in the management of SCA, reported significant anti-sickling activity in vitro [173].
5.2. Mushroom Nutritional Prospects
The typical herbal remedies used by traditional healers in different parts of Africa and elsewhere for the treatment of SCD were recently evaluated [174][175]. Safe, effective, and inexpensive therapeutic agents are urgently needed in Africa, and SCD management can be considered in two main aspects: medical protection and the management of major complications, both of which can be complemented by nutritional approaches [176].
Exogenous food-derived microRNAs, obtained through cross-kingdom modulation, may enter the human host from mushrooms, marine algae, and herbal teas. These microRNAs may serve as alternative tactics for new therapeutic effects, enhancing the existence of microRNA interactions between the diet, host, and gut microbiota [177][178][179].
Several reviews have revealed a solid connection between oxidative stress, inflammation, the immune response, and the pathogenesis of SCD, enhancing the role of the dominant natural immune system [180][181].
Despite the use of medicinal plants for millennia, information on African mushrooms in healthcare is less abundant [182][183]. Edible medicinal mushrooms, whether in the form of biomass, extracts, or derivatives, are potential sources of many bioactive products that can regulate immunity [184]. Mushroom bioactive elements exhibit various pharmacological activities, including multitargeting bioactivities, low toxicity, high safety, and affordability, making them valuable medicinal sources [185].
These fungal biochemical compounds include carbohydrates (β-glucans/lentinan, trehalose, chitosans) [185], carbohydrate-binding proteins (lectins) [186], mono- and polyunsaturated fatty acids(linoleic, oleic, palmitic) [187], phenolic compounds (caffeic, gallic, cinnamic, p-hydroxybenzoic, p-coumaric, and melatonin) [188], indole compounds (L-trytptophan) [189], vitamins (vitamins B complex, vitamin C, and tocopherols), terpenoids (carotenoids such as β-carotene and lycopene) [190], and unique molecules (e.g. ergothioneine and glutathione) [191][192].
Natural products, which have the ability to affect numerous targets and impact several signaling pathways, are widely recognized for their health benefits. Natural antioxidant-rich foods include berries, avocado, apples, cruciferous vegetables, nuts, olive oil, pulses, tomatoes, and mushrooms [193]. Mushrooms also possess strong anti-inflammatory effects. Despite being underappreciated as a medicinal source in Western countries, there is a growing need for further investigation, including their potential role in complementary prophylactic dietary treatments for sickle cell disease.
The consumption of edible mushrooms, either unprocessed or as supplements (extracts or as biomass), has been well-documented for many years as powerful instrument in maintaining health, longevity, and quality of life [194]. Their mode of action has been studied and can be attributed to their unique composition, including the essential amino acid (ergothioneine) [195], a type of β-glucans [196][197], specific enzymes, and secondary metabolites [198][199]. The way in which mushroom bioactive components interact with gut microbiota, influencing metabolism and various health disorders, has been the subject of many recent reviews [200][201].
Certain mushrooms are very rich in superoxide-dismutase, glutathione-peroxidase, catalase, and proteases, which are known to interact with transcription factors (e.g., nuclear-factor erythroid 2-related factor 2), thus preserving redox homeostasis in the cell and counteracting oxidative stress [202].
The regulatory effects of mushroom active ingredients (e.g., polyphenols) on ferroptosis has been described [203]. Edible mushrooms contain the ferroptosis inhibitor gallic acid, a natural hydroxybenzoic acid, which is also present in various foods such as nuts, red fruits, olive oil, green tea, and vegetables [204]. Ferroptocide is an inhibitor of thioredoxin, a central redox system in mammalian cells, that induces ferroptosis, which is the unique form of programmed death distinct from apoptosis, and maintains redox balances in sickle red blood cells [205][206][207].
Substantial oxidative stress is a prominent contributor to SCD due to a disproportionate yield of reactive oxygen species compared with the ability of antioxidant agents, including enzymes such as SODs, catalase, and glutathione peroxidase, to counteract them. Multiple inflammatory pathways are stimulated in a chain reaction involving sickle cell redox balance, hemolysis, vasculopathy, and regulation in the immune response [180].
Research on Ganoderma lucidum mushroom extracts revealed a significant decrease in hemoglobin polymerization rate, enhancing the binding of oxygen, thus preserving or stabilizing the structure of hemoglobin [208].
The mushroom Auricularia auricular, which is known for its medicinal properties [209] and anti-sickling potential, was recently studied in Nigeria. The study showed that this functional food, which has strong free radical scavenging activity, may reverse and stabilize erythrocyte membrane integrity and morphology. This means that this mushroom may offer a valuable natural option for the treatment and management of sickle cell anemia [210].
The mushroom Hericium erinaceus, mainly through its polysaccharide components, has traditionally and historically been used as a natural remedy for gastrointestinal disorders and epigastric pain.
Termite mushrooms (Termitomyces), a species from Africa and Asia, is used to increase hemoglobin levels (12.2 g/dL) and white blood cells (26,300 cells/mm3) [211] and contributes widely to modern medical research because its mycelial biomass expresses strong antioxidant potential [212] (Table 4).
Table 4. Mushrooms in sickle cell disease.
This entry is adapted from the peer-reviewed paper 10.3390/nu16020258
References
- Elendu, C.; Amaechi, D.C.; Alakwe-Ojimba, C.E.; Elendu, T.C.; Elendu, R.C.; Ayabazu, C.P.; Aina, T.O.; Aborisade, O.; Adenikinju, J.S. Understanding Sickle Cell Disease: Causes, Symptoms, and Treatment Options. Medicine 2023, 102, E35237.
- Tebbi, C.K. Sickle Cell Disease, a Review. Hemato 2022, 3, 341–366.
- Quinn, C.T. Minireview: Clinical Severity in Sickle Cell Disease: The Challenges of Definition and Prognostication. Exp. Biol. Med. 2016, 241, 679–688.
- Ershler, W.B.; De Castro, L.M.; Pakbaz, Z.; Moynahan, A.; Weycker, D.; Delea, T.E.; Agodoa, I.; Cong, Z. Hemoglobin and End-Organ Damage in Individuals with Sickle Cell Disease. Curr. Ther. Res. 2023, 98, 100696.
- Inusa, B.P.D.; Hsu, L.L.; Kohli, N.; Patel, A.; Ominu-Evbota, K.; Anie, K.A.; Atoyebi, W. Sickle Cell Disease—Genetics, Pathophysiology, Clinical Presentation and Treatment. Int. J. Neonatal Screen. 2019, 5, 20.
- Kargutkar, N.; Sawant-Mulay, M.; Hariharan, P.; Chandrakala, S.; Nadkarni, A. Role of MicroRNA in Hydroxyurea Mediated HbF Induction in Sickle Cell Anaemia Patients. Sci. Rep. 2023, 13, 369.
- Thom, C.S.; Dickson, C.F.; Gell, D.A.; Weiss, M.J. Hemoglobin Variants: Biochemical Properties and Clinical Correlates. Cold Spring Harb. Perspect. Med. 2013, 3, a011858.
- Aldakeel, S.A.; Ghanem, N.Z.; Al-Amodi, A.M.; Osman, A.K.; Al Asoom, L.I.; Ahmed, N.R.; Almandil, N.B.; Akhtar, M.S.; Azeez, S.A.; Borgio, J.F. Identification of Seven Novel Variants in the β-Globin Gene in Transfusion-Dependent and Normal Patients. Arch. Med. Sci. 2020, 16, 453–459.
- Egesa, W.I.; Nakalema, G.; Waibi, W.M.; Turyasiima, M.; Amuje, E.; Kiconco, G.; Odoch, S.; Kumbakulu, P.K.; Abdirashid, S.; Asiimwe, D. Sickle Cell Disease in Children and Adolescents: A Review of the Historical, Clinical, and Public Health Perspective of Sub-Saharan Africa and Beyond. Int. J. Pediatr. 2022, 2022, 3885979.
- Uçucu, S.; Karabıyık, T.; Azik, F. Difficulties in the Diagnosis of HbS/Beta Thalassemia: Really a Mild Disease? J. Med. Biochem. 2022, 41, 32–39.
- Liu, Y.; Su, S.; Shayo, S.; Bao, W.; Pal, M.; Dou, K.; Shi, P.A.; Aygun, B.; Campbell-Lee, S.; Lobo, C.A.; et al. Hemolysis Dictates Monocyte Differentiation via Two Distinct Pathways in Sickle Cell Disease Vaso-Occlusion. J. Clin. Investig. 2023, 133, e172087.
- Nader, E.; Romana, M.; Connes, P. The Red Blood Cell—Inflammation Vicious Circle in Sickle Cell Disease. Front. Immunol. 2020, 11, 517556.
- Mangla, A.; Ehsan, M.; Agarwal, N.; Maruvada, S. Sickle Cell Anemia. In StatPearls ; StatPearls Publishing: Treasure Island, FL, USA, 2023.
- Venugopal, A.; Chandran, M.; Eruppakotte, N.; Kizhakkillach, S.; Breezevilla, S.C.; Vellingiri, B. Monogenic Diseases in India. Mutat. Res. Mutat. Res. 2018, 776, 23–31.
- Mehta, A.B.; Hoffbrand, A.V. Hemolytic Anaemias V Inherited Defects of Haemoglobin—Sickle Cell Disease. In Haematology at a Glance; Blackwell Science: Malden, MA, USA, 2000; ISBN 9780632047932.
- Stuart, M.J.; Nagel, R.L. Sickle-Cell Disease. Lancet 2004, 364, 1343–1360.
- Habara, A.; Steinberg, M.H. Minireview: Genetic Basis of Heterogeneity and Severity in Sickle Cell Disease. Exp. Biol. Med. 2016, 241, 689–696.
- Du, M.; Van Ness, S.; Gordeuk, V.; Nouraie, S.M.; Nekhai, S.; Gladwin, M.; Steinberg, M.H.; Sebastiani, P. Biomarker Signatures of Sickle Cell Disease Severity. Blood Cells Mol. Dis. 2018, 72, 1–9.
- Njoku, F.; Zhang, X.; Shah, B.N.; Machado, R.F.; Han, J.; Saraf, S.L.; Gordeuk, V.R. Biomarkers of Clinical Severity in Treated and Untreated Sickle Cell Disease: A Comparison by Genotypes of a Single Center Cohort and African Americans in the NHANES Study. Br. J. Haematol. 2021, 194, 767–778.
- Takaoka, K.; Cyril, A.C.; Jinesh, S.; Radhakrishnan, R. Mechanisms of Pain in Sickle Cell Disease. Br. J. Pain 2020, 15, 213–220.
- Sadler, K.E.; Atkinson, S.N.; Ehlers, V.L.; Waltz, T.B.; Hayward, M.; García, D.M.R.; Salzman, N.H.; Stucky, C.L.; Brandow, A.M. Gut Microbiota and Metabolites Drive Chronic Sickle Cell Disease Pain. bioRxiv 2023.
- Matthie, N.; Ross, D.; Sinha, C.; Khemani, K.; Bakshi, N.; Krishnamurti, L. A Qualitative Study of Chronic Pain and Self-Management in Adults with Sickle Cell Disease. J. Natl. Med. Assoc. 2019, 111, 158–168.
- Osunkwo, I.; O’Connor, H.F.; Saah, E. Optimizing the Management of Chronic Pain in Sickle Cell Disease. Hematology 2020, 2020, 562–569.
- Thomson, A.M.; McHugh, T.A.; Oron, A.P.; Teply, C.; Lonberg, N.; Vilchis Tella, V.; Wilner, L.B.; Fuller, K.; Hagins, H.; Aboagye, R.G.; et al. Global, Regional, and National Prevalence and Mortality Burden of Sickle Cell Disease, 2000–2021: A Systematic Analysis from the Global Burden of Disease Study 2021. Lancet Haematol. 2023, 10, e585–e599.
- Grosse, S.D.; Odame, I.; Atrash, H.K.; Amendah, D.D.; Piel, F.B.; Williams, T.N. Sickle Cell Disease in Africa: A Neglected Cause of Early Childhood Mortality. Am. J. Prev. Med. 2011, 41, S398–S405.
- Delgadinho, M.; Ginete, C.; Santos, B.; Mendes, J.; Miranda, A.; Vasconcelos, J.; Brito, M. Microbial Gut Evaluation in an Angolan Paediatric Population with Sickle Cell Disease. J. Cell. Mol. Med. 2022, 26, 5360–5368.
- Ranque, B.; Kitenge, R.; Ndiaye, D.D.; Ba, M.D.; Adjoumani, L.; Traore, H.; Coulibaly, C.; Guindo, A.; Boidy, K.; Mbuyi, D.; et al. Estimating the Risk of Child Mortality Attributable to Sickle Cell Anaemia in Sub-Saharan Africa: A Retrospective, Multicentre, Case-Control Study. Lancet Haematol. 2022, 9, e208–e216.
- Ali Hazzazi, A.; Ageeli, M.H.; Alfaqih, A.M.; Ali Jaafari, A.; Malhan, H.M.; Bakkar, M.M. Epidemiology and Characteristics of Sickle Cell Patients Admitted to Hospitals in Jazan Region, Saudi Arabia. J. Appl. Hematol. 2020, 11, 10–14.
- Arji, E.E.; Eze, U.J.; Ezenwaka, G.O.; Kennedy, N. Evidence-Based Interventions for Reducing Sickle Cell Disease-Associated Morbidity and Mortality in Sub-Saharan Africa: A Scoping Review. SAGE Open Med. 2023, 11.
- Piel, F.B.; Rees, D.C.; DeBaun, M.R.; Nnodu, O.; Ranque, B.; Thompson, A.A.; Ware, R.E.; Abboud, M.R.; Abraham, A.; Ambrose, E.E.; et al. Defining Global Strategies to Improve Outcomes in Sickle Cell Disease: A Lancet Haematology Commission. Lancet Haematol. 2023, 10, e633–e686.
- Sedrak, A.; Kondamudi, N.P. Sickle Cell Disease. In StatPearls ; StatPearls Publishing: Treasure Island, FL, USA, 2023.
- Piel, F.B.; Hay, S.I.; Gupta, S.; Weatherall, D.J.; Williams, T.N. Global Burden of Sickle Cell Anaemia in Children under Five, 2010–2050: Modelling Based on Demographics, Excess Mortality, and Interventions. PLoS Med. 2013, 10, e1001484.
- Ansong, D.; Akoto, A.O.; Ocloo, D.; Ohene-Frempong, K. Sickle Cell Disease: Management Options and Challenges in Developing Countries. Mediterr. J. Hematol. Infect. Dis. 2013, 5, e2013062.
- Colombatti, R.; Hegemann, I.; Medici, M.; Birkegård, C. Systematic Literature Review Shows Gaps in Data on Global Prevalence and Birth Prevalence of Sickle Cell Disease and Sickle Cell Trait: Call for Action to Scale up and Harmonize Data Collection. J. Clin. Med. 2023, 12, 5538.
- Adigwe, O.P.; Onoja, S.O.; Onavbavba, G. A Critical Review of Sickle Cell Disease Burden and Challenges in Sub-Saharan Africa. J. Blood Med. 2023, 14, 367–376.
- Piel, F.B.; Patil, A.P.; Howes, R.E.; Nyangiri, O.A.; Gething, P.W.; Dewi, M.; Temperley, W.H.; Williams, T.N.; Weatherall, D.J.; Hay, S.I. Global Epidemiology of Sickle Haemoglobin in Neonates: A Contemporary Geostatistical Model-Based Map and Population Estimates. Lancet 2013, 381, 142–151.
- Nieuwenhuis, F.; Wolf, B.; Bomba, A.; Graaf, P. De Haematological Study in Cabo Delgado Province, Mozambique; Sickle Cell Trait and G6PD Deficiency. Trop. Geogr. Med. 1986, 38, 183–187.
- Galatas, B.; Mabote, L.; Simone, W.; Matambisso, G.; Nhamussua, L.; Mañú-Pereira, M.D.M.; Menéndez, C.; Saute, F.; Macete, E.; Bassat, Q.; et al. Heterogeneity of G6PD Deficiency Prevalence in Mozambique: A School-Based Cross-Sectional Survey in Three Different Regions CIBS-25/013 CIBS IRB00002657 IRB. Malar. J. 2017, 16, 36.
- Williams, T.N.; Mwangi, T.W.; Wambua, S.; Alexander, N.D.; Kortok, M.; Snow, R.W.; Marsh, K. Sickle Cell Trait and the Risk of Plasmodium Falciparum Malaria and Other Childhood Diseases. J. Infect. Dis. 2005, 192, 178–186.
- Saelens, J.W.; Petersen, J.E.V.; Freedman, E.; Moseley, R.C.; Konaté, D.; Diakité, S.A.S.; Traoré, K.; Vance, N.; Fairhurst, R.M.; Diakité, M.; et al. Impact of Sickle Cell Trait Hemoglobin on the Intraerythrocytic Transcriptional Program of Plasmodium Falciparum. mSphere 2021, 6, e0075521.
- Ngou, C.M.; Bayibéki, A.N.; Abate, L.; Makinde, O.S.; Feufack-Donfack, L.B.; Sarah-Matio, E.M.; Bouopda-Tuedom, A.G.; Taconet, P.; Moiroux, N.; Awono-Ambéné, P.H.; et al. Influence of the Sickle Cell Trait on Plasmodium Falciparum Infectivity from Naturally Infected Gametocyte Carriers. BMC Infect. Dis. 2023, 23, 317.
- Sesethu, G.; Nombalentle, M.; Yamkela, M.; Anelisa, M.; Makumire, S.; Mkwetshana, N.; Govender, K.K.; Makhoba, X.H. In Silico Evaluation of Heat Shock Proteins Reveals an Interplay with Polyamines as a Survival Strategy for the Plasmodium falciparum. INNOSC Theranostics Pharmacol. Sci. 2023, 1228.
- Aolymat, I.; Hatmal, M.M.; Olaimat, A.N. The Emerging Role of Heat Shock Factor 1 (HSF1) and Heat Shock Proteins (HSPs) in Ferroptosis. Pathophysiology 2023, 30, 63–82.
- Consoli, V.; Sorrenti, V.; Grosso, S.; Vanella, L. Heme Oxygenase-1 Signaling and Redox Homeostasis in Physiopathological Conditions. Biomolecules 2021, 11, 589.
- Menon, A.V.; Liu, J.; Tsai, H.P.; Zeng, L.; Yang, S.; Asnani, A.; Kim, J. Excess Heme Upregulates Heme Oxygenase 1 and Promotes Cardiac Ferroptosis in Mice with Sickle Cell Disease. Blood 2022, 139, 936–941.
- Rutter, M. Gene–Environment Interdependence. Dev. Sci. 2007, 10, 12–18.
- Bodaghi, A.; Fattahi, N.; Ramazani, A. Biomarkers: Promising and Valuable Tools towards Diagnosis, Prognosis and Treatment of COVID-19 and Other Diseases. Heliyon 2023, 9, e13323.
- Kavanagh, P.L.; Fasipe, T.A.; Wun, T. Sickle Cell Disease: A Review. JAMA 2022, 328, 57–68.
- Matter, B.; Seiler, C.L.; Murphy, K.; Ming, X.; Zhao, J.; Lindgren, B.; Jones, R.; Tretyakova, N. Mapping Three Guanine Oxidation Products along DNA Following Exposure to Three Types of Reactive Oxygen Species. Free Radic. Biol. Med. 2018, 121, 180–189.
- Balanikas, E.; Banyasz, A.; Baldacchino, G.; Markovitsi, D. Populations and Dynamics of Guanine Radicals in DNA Strands—Direct versus Indirect Generation. Molecules 2019, 24, 2347.
- Sesti-Costa, R.; Costa, F.F.; Conran, N. Role of Macrophages in Sickle Cell Disease Erythrophagocytosis and Erythropoiesis. Int. J. Mol. Sci. 2023, 24, 6333.
- Jamwal, S.; Blackburn, J.K.; Elsworth, J.D. PPARγ/PGC1α Signaling as a Potential Therapeutic Target for Mitochondrial Biogenesis in Neurodegenerative Disorders. Pharmacol. Ther. 2021, 219, 107705.
- Mihaylov, S.R.; Castelli, L.M.; Lin, Y.H.; Gül, A.; Soni, N.; Hastings, C.; Flynn, H.R.; Păun, O.; Dickman, M.J.; Snijders, A.P.; et al. The Master Energy Homeostasis Regulator PGC-1α Exhibits an MRNA Nuclear Export Function. Nat. Commun. 2023, 14, 5496.
- Vasquez-Martinez, G.; Mayoral-Andrade, G.; Tomanek-Chalkley, A.; Zepeda-Orozco, D.; Allen, B.G.; Mapuskar, K.A.; Vasquez-Martinez, G.; Mayoral-Andrade, G.; Tomanek-Chalkley, A.; Zepeda-Orozco, D.; et al. Mitochondrial Oxidative Metabolism: An Emerging Therapeutic Target to Improve CKD Outcomes. Biomedicines 2023, 11, 1573.
- Williams, T.N.; Thein, S.L. Sickle Cell Anemia and Its Phenotypes. Annu. Rev. Genom. Hum. Genet. 2018, 19, 113–147.
- Arishi, W.A.; Al-hadrami, H.A.; Zourob, M. Techniques for the Detection of Sickle Cell Disease: A Review. Micromachines 2021, 12, 519.
- Ontario Health (Quality). Carrier Screening Programs for Cystic Fibrosis, Fragile X Syndrome, Hemoglobinopathies and Thalassemia, and Spinal Muscular Atrophy: A Health Technology Assessment. Ont. Health Technol. Assess. Ser. 2023, 23, 1–398.
- Yenilmez, E.D.; Tuli, A. New Perspectives in Prenatal Diagnosis of Sickle Cell Anemia. In Sickle Cell Disease Pain and Common Chronic Complications; Inusa, B.P.D., Ed.; IntechOpen: London, UK, 2016; ISBN 978-953-51-2767-3.
- Bou-Fakhredin, R.; De Franceschi, L.; Motta, I.; Cappellini, M.D.; Taher, A.T. Pharmacological Induction of Fetal Hemoglobin in β-Thalassemia and Sickle Cell Disease: An Updated Perspective. Pharmaceuticals 2022, 15, 753.
- Hassan Murad, M.; Liem, R.I.; Lang, E.S.; Akl, E.A.; Meerpohl, J.J.; DeBaun, M.R.; Tisdale, J.F.; Brandow, A.M.; Lanzkron, S.M.; Chou, S.T.; et al. 2019 Sickle Cell Disease Guidelines by the American Society of Hematology: Methodology, Challenges, and Innovations. Blood Adv. 2019, 3, 3945–3950.
- Brandow, A.M.; Carroll, C.P.; Creary, S.; Edwards-Elliott, R.; Glassberg, J.; Hurley, R.W.; Kutlar, A.; Seisa, M.; Stinson, J.; Strouse, J.J.; et al. American Society of Hematology 2020 Guidelines for Sickle Cell Disease: Management of Acute and Chronic Pain. Blood Adv. 2020, 4, 2656–2701.
- Ali, M.A.; Ahmad, A.; Chaudry, H.; Aiman, W.; Aamir, S.; Anwar, M.Y.; Khan, A. Efficacy and Safety of Recently Approved Drugs for Sickle Cell Disease: A Review of Clinical Trials. Exp. Hematol. 2020, 92, 11–18.e1.
- Ware, R.E. Optimizing Hydroxyurea Therapy for Sickle Cell Anemia. Hematology 2015, 2015, 436–443.
- EMA. Oxbryta: EPAR—Medicine Overview; EMA/131392/2022; EMA: Amsterdam, The Netherlands, 2022.
- Eaton, W.A.; Bunn, H.F. Treating Sickle Cell Disease by Targeting HbS Polymerization. Blood 2017, 129, 2719–2726.
- Darshana, T.; Rees, D.; Premawardhena, A. Hydroxyurea and Blood Transfusion Therapy for Sickle Cell Disease in South Asia: Inconsistent Treatment of a Neglected Disease. Orphanet J. Rare Dis. 2021, 16, 148.
- Ferreira de Matos, C.; Comont, T.; Castex, M.P.; Lafaurie, M.; Walter, O.; Moulis, G.; Dion, J.; Cougoul, P. Risk of Vaso-Occlusive Episodes in Patients with Sickle Cell Disease Exposed to Systemic Corticosteroids: A Comprehensive Review. Expert Rev. Hematol. 2022, 15, 1045–1054.
- Telen, M.J.; Wun, T.; McCavit, T.L.; De Castro, L.M.; Krishnamurti, L.; Lanzkron, S.; Hsu, L.L.; Smith, W.R.; Rhee, S.; Magnani, J.L.; et al. Randomized Phase 2 Study of GMI-1070 in SCD: Reduction in Time to Resolution of Vaso-Occlusive Events and Decreased Opioid Use. Blood 2015, 125, 2656–2664.
- Doss, J.F.; Jonassaint, J.C.; Garrett, M.E.; Ashley-Koch, A.E.; Telen, M.J.; Chi, J.T. Phase 1 Study of a Sulforaphane-Containing Broccoli Sprout Homogenate for Sickle Cell Disease. PLoS ONE 2016, 11, e0152895.
- Khaddour, K.; Hana, C.K.; Mewawalla, P. Hematopoietic Stem Cell Transplantation. In StatPearls ; StatPearls Publishing: Treasure Island, FL, USA, 2023.
- Chou, S.T.; Alsawas, M.; Fasano, R.M.; Field, J.J.; Hendrickson, J.E.; Howard, J.; Kameka, M.; Kwiatkowski, J.L.; Pirenne, F.; Shi, P.A.; et al. American Society of Hematology 2020 Guidelines for Sickle Cell Disease: Transfusion Support. Blood Adv. 2020, 4, 327–355.
- Han, H.; Hensch, L.; Tubman, V.N. Indications for Transfusion in the Management of Sickle Cell Disease. Hematology 2021, 2021, 696–703.
- Rankine-Mullings, A.E.; Nevitt, S.J. Hydroxyurea (Hydroxycarbamide) for Sickle Cell Disease. Cochrane Database Syst. Rev. 2022, 2022, CD002202.
- Inam, Z.; Tisdale, J.F.; Leonard, A. Outcomes and Long-Term Effects of Hematopoietic Stem Cell Transplant in Sickle Cell Disease. Expert Rev. Hematol. 2023, 16, 879–903.
- Bhalla, N.; Bhargav, A.; Yadav, S.K.; Singh, A.K. Allogeneic Hematopoietic Stem Cell Transplantation to Cure Sickle Cell Disease: A Review. Front. Med. 2023, 10, 1036939.
- Sadaf, A.; Quinn, C.T. L-Glutamine for Sickle Cell Disease: Knight or Pawn? Exp. Biol. Med. 2020, 245, 146–154.
- Yenamandra, A.; Marjoncu, D. Voxelotor: A Hemoglobin S Polymerization Inhibitor for the Treatment of Sickle Cell Disease. J. Adv. Pract. Oncol. 2020, 11, 873–877.
- Karki, N.R.; Saunders, K.; Kutlar, A. A Critical Evaluation of Crizanlizumab for the Treatment of Sickle Cell Disease. Expert Rev. Hematol. 2022, 15, 5–13.
- Gov. UK. MHRA Authorises World-First Gene Therapy That Aims to Cure Sickle-Cell Disease and Transfusion-Dependent β-Thalassemia. Available online: https://www.gov.uk/government/news/mhra-authorises-world-first-gene-therapy-that-aims-to-cure-sickle-cell-disease-and-transfusion-dependent-thalassemia (accessed on 31 December 2023).
- De Souza, D.C.; Hebert, N.; Esrick, E.B.; Ciuculescu, M.F.; Archer, N.M.; Armant, M.; Audureau, É.; Brendel, C.; Di Caprio, G.; Galactéros, F.; et al. Genetic Reversal of the Globin Switch Concurrently Modulates Both Fetal and Sickle Hemoglobin and Reduces Red Cell Sickling. Nat. Commun. 2023, 14, 5850.
- The National Heart, Lung, and Blood Institute (NHLBI). The Management of Sickle Cell Disease; NIH Publication: Bethesda, MD, USA, 2002.
- Wang, W.C. Sickle Cell Anemia and Other Sickling Syndromes. In Wintrobe’s Clinical Hematology; Greer, J.P., Foerster, J., Rodgers, G.M., Paraskevas, F., Glader, B., Arber, D.A., Means, R.T., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2009; ISBN 978-0781765077.
- Ratko, T.A.; Belinson, S.E.; Brown, H.M.; Noorani, H.Z.; Chopra, R.D.; Marbella, A.; Samson, D.J.; Bonnell, C.J.; Ziegler, K.M.; Aronson, N. Hematopoietic Stem-Cell Transplantation in the Pediatric Population; AHRO: Rockville, MD, USA, 2012.
- Gee, B.E. Biologic Complexity in Sickle Cell Disease: Implications for Developing Targeted Therapeutics. Sci. World J. 2013, 2013, 694146.
- ABScience. Available online: https://www.ab-science.com/news-and-media/press-releases/ (accessed on 31 December 2023).
- Conran, N.; Belcher, J.D. Inflammation in Sickle Cell Disease. Clin. Hemorheol. Microcirc. 2018, 68, 263–299.
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between Microbiota and Immunity in Health and Disease. Cell Res. 2020, 30, 492–506.
- Zhao, L.Y.; Mei, J.X.; Yu, G.; Lei, L.; Zhang, W.H.; Liu, K.; Chen, X.L.; Kołat, D.; Yang, K.; Hu, J.K. Role of the Gut Microbiota in Anticancer Therapy: From Molecular Mechanisms to Clinical Applications. Signal Transduct. Target. Ther. 2023, 8, 201.
- Thursby, E.; Juge, N. Introduction to the Human Gut Microbiota. Biochem. J. 2017, 474, 1823–1836.
- Cabrera-Mulero, A.; Tinahones, A.; Bandera, B.; Moreno-Indias, I.; Macías-González, M.; Tinahones, F.J. Keto Microbiota: A Powerful Contributor to Host Disease Recovery. Rev. Endocr. Metab. Disord. 2019, 20, 415–425.
- Roy, S.; Nag, S.; Saini, A.; Choudhury, L. Association of Human Gut Microbiota with Rare Diseases: A Close Peep Through. Intractable Rare Dis. Res. 2022, 11, 52–62.
- Brim, H.; Taylor, J.; Abbas, M.; Vilmenay, K.; Daremipouran, M.; Varma, S.; Lee, E.; Pace, B.; Song-Naba, W.L.; Gupta, K.; et al. The Gut Microbiome in Sickle Cell Disease: Characterization and Potential Implications. PLoS ONE 2021, 16, e0255956.
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-Gut Microbiota Metabolic Interactions. Science 2012, 336, 1262–1267.
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment Dominates over Host Genetics in Shaping Human Gut Microbiota. Nature 2018, 555, 210–215.
- Zhang, H.; Sparks, J.B.; Karyala, S.V.; Settlage, R.; Luo, X.M. Host Adaptive Immunity Alters Gut Microbiota. ISME J. 2014, 9, 770–781.
- Dike, C.R.; Hanson, C.; Davies, H.D.; Obaro, S.; Yu, F.; Harper, J.; Grace, H.; Lebensburger, J.; Raulji, C.; Ma, J.; et al. The Relationship between Nutrition, Gut Dysbiosis, and Pediatric Sickle Cell Pain Outcomes: A Pilot Study. Pediatr. Blood Cancer 2023, 70, e30397.
- Mallott, E.K.; Sitarik, A.R.; Leve, L.D.; Cioffi, C.; Camargo, C.A.; Hasegawa, K.; Bordenstein, S.R. Human Microbiome Variation Associated with Race and Ethnicity Emerges as Early as 3 Months of Age. PLoS Biol. 2023, 21, e3002230.
- Qin, Y.; Havulinna, A.S.; Liu, Y.; Jousilahti, P.; Ritchie, S.C.; Tokolyi, A.; Sanders, J.G.; Valsta, L.; Brożyńska, M.; Zhu, Q.; et al. Combined Effects of Host Genetics and Diet on Human Gut Microbiota and Incident Disease in a Single Population Cohort. Nat. Genet. 2022, 54, 134–142.
- Malard, F.; Dore, J.; Gaugler, B.; Mohty, M. Introduction to Host Microbiome Symbiosis in Health and Disease. Mucosal Immunol. 2021, 14, 547–554.
- Mutalub, Y.B.; Abdulwahab, M.; Mohammed, A.; Yahkub, A.M.; AL-Mhanna, S.B.; Yusof, W.; Tang, S.P.; Rasool, A.H.G.; Mokhtar, S.S. Gut Microbiota Modulation as a Novel Therapeutic Strategy in Cardiometabolic Diseases. Foods 2022, 11, 2575.
- Huda, M.N.; Salvador, A.C.; Barrington, W.T.; Gacasan, C.A.; D’Souza, E.M.; Deus Ramirez, L.; Threadgill, D.W.; Bennett, B.J. Gut Microbiota and Host Genetics Modulate the Effect of Diverse Diet Patterns on Metabolic Health. Front. Nutr. 2022, 9, 896348.
- Lajqi, T.; Pöschl, J.; Frommhold, D.; Hudalla, H. The Role of Microbiota in Neutrophil Regulation and Adaptation in Newborns. Front. Immunol. 2020, 11, 568685.
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T.; et al. Human Genetics Shape the Gut Microbiome. Cell 2014, 159, 789–799.
- Turpin, W.; Espin-Garcia, O.; Xu, W.; Silverberg, M.S.; Kevans, D.; Smith, M.I.; Guttman, D.S.; Griffiths, A.; Panaccione, R.; Otley, A.; et al. Association of Host Genome with Intestinal Microbial Composition in a Large Healthy Cohort. Nat. Genet. 2016, 48, 1413–1417.
- Blouin, M.J.; Beauchemin, H.; Wright, A.; De Paepe, M.; Soretie, M.; Bleau, A.M.; Nakamoto, B.; Ou, C.N.; Stamatoyannopoulos, G.; Trudel, M. Genetic Correction of Sickle Cell Disease: Insights Using Transgenic Mouse Models. Nat. Med. 2000, 6, 177–182.
- Beuzard, Y. Mouse Models of Sickle Cell Disease. Transfus. Clin. Biol. 2008, 15, 7–11.
- Pawliuk, R.; Westerman, K.A.; Fabry, M.E.; Payen, E.; Tighe, R.; Bouhassira, E.E.; Acharya, S.A.; Ellis, J.; London, I.M.; Eaves, C.J.; et al. Correction of Sickle Cell Disease in Transgenic Mouse Models by Gene Therapy. Science 2001, 294, 2368–2371.
- Samakoglu, S.; Lisowski, L.; Budak-Alpdogan, T.; Usachenko, Y.; Acuto, S.; Di Marzo, R.; Maggio, A.; Zhu, P.; Tisdale, J.F.; Riviere, I.; et al. A Genetic Strategy to Treat Sickle Cell Anemia by Coregulating Globin Transgene Expression and RNA Interference. Nat. Biotechnol. 2005, 24, 89–94.
- Lewis, C.V.; Sellak, H.; Sawan, M.A.; Joseph, G.; Darby, T.M.; VanInsberghe, D.; Naudin, C.R.; Archer, D.R.; Jones, R.M.; Taylor, W.R. Intestinal Barrier Dysfunction in Murine Sickle Cell Disease Is Associated with Small Intestine Neutrophilic Inflammation, Oxidative Stress, and Dysbiosis. FASEB BioAdvances 2023, 5, 199–210.
- Forcados, G.E.; Muhammad, A.; Oladipo, O.O.; Makama, S.; Meseko, C.A. Metabolic Implications of Oxidative Stress and Inflammatory Process in SARS-CoV-2 Pathogenesis: Therapeutic Potential of Natural Antioxidants. Front. Cell. Infect. Microbiol. 2021, 11, 654813.
- Martemucci, G.; Costagliola, C.; Mariano, M.; D’andrea, L.; Napolitano, P.; D’Alessandro, A.G. Free Radical Properties, Source and Targets, Antioxidant Consumption and Health. Oxygen 2022, 2, 48–78.
- Ren, H.; Ghebremeskel, K.; Okpala, I.; Lee, A.; Ibegbulam, O.; Crawford, M. Patients with Sickle Cell Disease Have Reduced Blood Antioxidant Protection. Int. J. Vitam. Nutr. Res. 2013, 78, 139–147.
- Setty, B.N.Y.; Betal, S.G.; Miller, R.E.; Brown, D.S.; Meier, M.; Cahill, M.; Lerner, N.B.; Apollonsky, N.; Stuart, M.J. Relationship of Omega-3 Fatty Acids DHA and EPA with the Inflammatory Biomarker Hs-CRP in Children with Sickle Cell Anemia. Prostaglandins Leukot. Essent. Fat. Acids 2019, 146, 11–18.
- Daak, A.A.; Lopez-Toledano, M.A.; Heeney, M.M. Biochemical and Therapeutic Effects of Omega-3 Fatty Acids in Sickle Cell Disease. Complement. Ther. Med. 2020, 52, 102482.
- Pittman, D.D.; Hines, P.C.; Beidler, D.; Rybin, D.; Frelinger, A.L.; Michelson, A.D.; Liu, K.; Gao, X.; White, J.; Zaidi, A.U.; et al. Evaluation of Longitudinal Pain Study in Sickle Cell Disease (ELIPSIS) by Patient-Reported Outcomes, Actigraphy, and Biomarkers. Blood 2021, 137, 2010–2020.
- Sadler, K.E.; Mogil, J.S.; Stucky, C.L. Innovations and Advances in Modelling and Measuring Pain in Animals. Nat. Rev. Neurosci. 2021, 23, 70–85.
- Sadler, K.; Ehlers, V.; Brandow, A.; Stucky, C. Sickle Cell Disease Associated Changes in the Gut Microbiome Contribute to Persistent Pain. J. Pain 2022, 23, 7.
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141.
- Xu, F.; Fu, Y.; Sun, T.Y.; Jiang, Z.; Miao, Z.; Shuai, M.; Gou, W.; Ling, C.W.; Yang, J.; Wang, J.; et al. The Interplay between Host Genetics and the Gut Microbiome Reveals Common and Distinct Microbiome Features for Complex Human Diseases. Microbiome 2020, 8, 145.
- Quan, Y.; Zhang, K.X.; Zhang, H.Y. The Gut Microbiota Links Disease to Human Genome Evolution. Trends Genet. 2023, 39, 451–461.
- Dutta, D.; Aujla, A.; Knoll, B.M.; Lim, S.H. Intestinal Pathophysiological and Microbial Changes in Sickle Cell Disease: Potential Targets for Therapeutic Intervention. Br. J. Haematol. 2020, 188, 488–493.
- Umeakunne, K.; Hibbert, J.M. Nutrition in Sickle Cell Disease: Recent Insights. Nutr. Diet. Suppl. 2019, 11, 9–17.
- Reber, E.; Gomes, F.; Vasiloglou, M.F.; Schuetz, P.; Stanga, Z. Nutritional Risk Screening and Assessment. J. Clin. Med. 2019, 8, 1065.
- Ohemeng, A.; Nartey, E.B.; Quaidoo, E.; Ansong, R.S.; Asiedu, M.S. Knowledge and Nutrition-Related Practices among Caregivers of Adolescents with Sickle Cell Disease in the Greater Accra Region of Ghana. BMC Public Health 2023, 23, 434.
- Boadu, I.; Ohemeng, A.; Renner, L.A. Dietary Intakes and Nutritional Status of Children with Sickle Cell Disease at the Princess Marie Louise Hospital, Accra—A Survey. BMC Nutr. 2018, 4, 33.
- Fernandes, T.H.; Bell, V. The Imprecision of Micronutrient Requirement Values: The Example of Vitamin D. J. Food Sci. 2023, 89, 51–63.
- Zemel, B.S.; Kawchak, D.A.; Fung, E.B.; Ohene-Frempong, K.; Stallings, V.A. Effect of Zinc Supplementation on Growth and Body Composition in Children with Sickle Cell Disease. Am. J. Clin. Nutr. 2002, 75, 300–307.
- Hibbert, J.M.; Hsu, L.L.; Bhathena, S.J.; Irune, I.; Sarfo, B.; Creary, M.S.; Gee, B.E.; Mohamed, A.I.; Buchanan, I.D.; Al-Mahmoud, A.; et al. Proinflammatory Cytokines and the Hypermetabolism of Children with Sickle Cell Disease. Exp. Biol. Med. 2005, 230, 68–74.
- McCormick, M.; Osei-Anto, H.A.; Martinez, R.M. Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action; National Academies Press: Washington, DC, USA, 2020.
- Dutta, D.; Methe, B.; Amar, S.; Morris, A.; Lim, S.H. Intestinal Injury and Gut Permeability in Sickle Cell Disease. J. Transl. Med. 2019, 17, 183.
- Haroun, E.; Kumar, P.A.; Saba, L.; Kassab, J.; Ghimire, K.; Dutta, D.; Lim, S.H. Intestinal Barrier Functions in Hematologic and Oncologic Diseases. J. Transl. Med. 2023, 21, 233.
- Hyacinth, H.I.; Gee, B.E.; Hibbert, J.M. The Role of Nutrition in Sickle Cell Disease. Nutr. Metab. Insights 2010, 3.
- Da Guarda, C.C.; Yahouédéhou, S.C.M.A.; Santiago, R.P.; Neres, J.S.D.S.; Fernandes, C.F.D.L.; Aleluia, M.M.; Figueiredo, C.V.B.; Fiuza, L.M.; Carvalho, S.P.; Oliveira, R.M.D.; et al. Sickle Cell Disease: A Distinction of Two Most Frequent Genotypes (HbSS and HbSC). PLoS ONE 2020, 15, e0228399.
- Soe, H.H.K.; Abas, A.B.L.; Than, N.N.; Ni, H.; Singh, J.; Said, A.R.B.M.; Osunkwo, I. Vitamin D Supplementation for Sickle Cell Disease. Cochrane Database Syst. Rev. 2020, 5, CD010858.
- Cairo, C.; Webb, T.J. Effective Barriers: The Role of NKT Cells and Innate Lymphoid Cells in the Gut. J. Immunol. 2022, 208, 235–246.
- Yin, R.; Kuo, H.C.; Hudlikar, R.; Sargsyan, D.; Li, S.; Wang, L.; Wu, R.; Kong, A.N. Gut Microbiota, Dietary Phytochemicals, and Benefits to Human Health. Curr. Pharmacol. Rep. 2019, 5, 332–344.
- Townsend, J.R.; Kirby, T.O.; Marshall, T.M.; Church, D.D.; Jajtner, A.R.; Esposito, R. Foundational Nutrition: Implications for Human Health. Nutrients 2023, 15, 2837.
- Teixeira, T.V.; Da Silva, A.C.F.; Rodrigues, C.d.S.C.; Brito, F.d.S.B.; Canella, D.S.; Citelli, M. Food Consumption of People with Sickle Cell Anemia in a Middle-Income Country. Nutrients 2023, 15, 1478.
- Al-Saqladi, A.W.M.; Cipolotti, R.; Fijnvandraat, K.; Brabin, B.J. Growth and Nutritional Status of Children with Homozygous Sickle Cell Disease. Ann. Trop. Paediatr. 2008, 28, 165–189.
- Bello-Manga, H.; DeBaun, M.R.; Kassim, A.A. Epidemiology and Treatment of Relative Anemia in Children with Sickle Cell Disease in Sub-Saharan Africa. Expert Rev. Hematol. 2016, 9, 1031–1042.
- Nartey, E.B.; Spector, J.; Adu-Afarwuah, S.; Jones, C.L.; Jackson, A.; Ohemeng, A.; Shah, R.; Koryo-Dabrah, A.; Kuma, A.B.A.; Hyacinth, H.I.; et al. Nutritional Perspectives on Sickle Cell Disease in Africa: A Systematic Review. BMC Nutr. 2021, 7, 9.
- Sahu, T.; Pande, B.; Verma, H.K.; Bhaskar, L.V.K.S.; Sinha, M.; Sinha, R.; Rao, P.V. Infection and Potential Challenge of Childhood Mortality in Sickle Cell Disease: A Comprehensive Review of the Literature from a Global Perspective. Thalass. Rep. 2023, 13, 206–229.
- Kamal, S.; Naghib, M.M.; Zahrani, J.A.; Hassan, H.; Moawad, K.; Arrahman, O. The Influence of Nutrition on Disease Severity and Health-Related Quality of Life in Adults with Sickle Cell Disease: A Prospective Longitudinal Study. Mediterr. J. Hematol. Infect. Dis. 2021, 13, e2021007.
- UNICEF. Fed to Fail? The Crisis of Children’s Diets in Early Life. 2021 Child Nutrition Report; UN Children’s Fund: New York, NY, USA, 2021.
- Cox, S.E.; Makani, J.; Fulford, A.J.; Komba, A.N.; Soka, D.; Williams, T.N.; Newton, C.R.; Marsh, K.; Prentice, A.M. Nutritional Status, Hospitalization and Mortality among Patients with Sickle Cell Anemia in Tanzania. Haematologica 2011, 96, 948–953.
- Danton, O.; Somboro, A.; Fofana, B.; Diallo, D.; Sidibé, L.; Rubat-Coudert, C.; Marchand, F.; Eschalier, A.; Ducki, S.; Chalard, P. Ethnopharmacological Survey of Plants Used in the Traditional Treatment of Pain Conditions in Mali. J. Herb. Med. 2019, 17–18, 100271.
- Delesderrier, E.; Curioni, C.; Omena, J.; Macedo, C.R.; Cople-Rodrigues, C.; Citelli, M. Antioxidant Nutrients and Hemolysis in Sickle Cell Disease. Clin. Chim. Acta 2020, 510, 381–390.
- Darbari, D.S.; Sheehan, V.A.; Ballas, S.K. The Vaso-Occlusive Pain Crisis in Sickle Cell Disease: Definition, Pathophysiology, and Management. Eur. J. Haematol. 2020, 105, 237–246.
- Reed, J.D.; Redding-Lallinger, R.; Orringer, E.P. Nutrition and Sickle Cell Disease. Am. J. Hematol. 1987, 24, 441–455.
- Dixit, R.; Nettem, S.; Madan, S.S.; Soe, H.H.K.; Abas, A.B.L.; Vance, L.D.; Stover, P.J. Folate Supplementation in People with Sickle Cell Disease. Cochrane Database Syst. Rev. 2018, 2018, CD011130.
- Arruda, M.M.; Mecabo, G.; Rodrigues, C.A.; Matsuda, S.S.; Rabelo, I.B.; Figueiredo, M.S. Antioxidant Vitamins C and E Supplementation Increases Markers of Haemolysis in Sickle Cell Anaemia Patients: A Randomized, Double-Blind, Placebo-Controlled Trial. Br. J. Haematol. 2013, 160, 688–700.
- Onalo, R.; Cooper, P.; Cilliers, A.; Vorster, B.C.; Uche, N.A.; Oluseyi, O.O.; Onalo, V.D.; Zubairu, Y.; Ayodele-Kehinde, A.U.; Damilare, O.M.; et al. Randomized Control Trial of Oral Arginine Therapy for Children with Sickle Cell Anemia Hospitalized for Pain in Nigeria. Am. J. Hematol. 2021, 96, 89–97.
- Sadeghi, A.; Taherifard, E.; Dehdari Ebrahimi, N.; Rafiei, E.; Hadianfard, F.; Taherifard, E. Effects of L-Arginine Supplementation in Patients with Sickle Cell Disease: A Systematic Review and Meta-Analysis of Clinical Trials. Health Sci. Reports 2023, 6, e1167.
- Arribas-López, E.; Zand, N.; Ojo, O.; Snowden, M.J.; Kochhar, T. The Effect of Amino Acids on Wound Healing: A Systematic Review and Meta-Analysis on Arginine and Glutamine. Nutrients 2021, 13, 2498.
- Biswal, S.; Rizwan, H.; Pal, S.; Sabnam, S.; Parida, P.; Pal, A. Oxidative Stress, Antioxidant Capacity, Biomolecule Damage, and Inflammation Symptoms of Sickle Cell Disease in Children. Hematology 2019, 24, 1–9.
- Dosunmu-Ogunbi, A.M.; Wood, K.C.; Novelli, E.M.; Straub, A.C. Decoding the Role of SOD2 in Sickle Cell Disease. Blood Adv. 2019, 3, 2679–2687.
- Forman, H.J.; Zhang, H. Targeting Oxidative Stress in Disease: Promise and Limitations of Antioxidant Therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709.
- Juan, C.A.; de la Lastra, J.M.P.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642.
- Enomoto, T.M.; Isichei, C.; Vanderjagt, D.J.; Fry, D.E.; Glew, R.H. Decreased Polyunsaturated Fatty Acids in Sickle Cell Anaemia. J. Trop. Pediatr. 1998, 44, 28–34.
- Lanza, V.; Greco, V.; Bocchieri, E.; Sciuto, S.; Inturri, R.; Messina, L.; Vaccaro, S.; Bellia, F.; Rizzarelli, E. Synergistic Effect of L-Carnosine and Hyaluronic Acid in Their Covalent Conjugates on the Antioxidant Abilities and the Mutual Defense against Enzymatic Degradation. Antioxidants 2022, 11, 664.
- Antwi-Boasiako, C.; Dankwah, G.B.; Aryee, R.; Hayfron-Benjamin, C.; Donkor, E.S.; Campbell, A.D. Oxidative Profile of Patients with Sickle Cell Disease. Med. Sci. 2019, 7, 17.
- Djordjević, V.V.; Kostić, J.; Krivokapić, Ž.; Krtinić, D.; Ranković, M.; Petković, M.; Ćosić, V. Decreased Activity of Erythrocyte Catalase and Glutathione Peroxidase in Patients with Schizophrenia. Medicina 2022, 58, 1491.
- WHO. Special Issue 14—African Traditional Medicine Day. The African Health Monitor; 2010. Available online: https://www.afro.who.int/sites/default/files/2017-06/ahm-special-issue-14.pdf (accessed on 31 December 2023).
- Mahomoodally, M.F. Traditional Medicines in Africa: An Appraisal of Ten Potent African Medicinal Plants. Evid.-Based Complement. Altern. Med. 2013, 2013, 617459.
- WHO. Sickle-Cell Anaemia; WHO: Geneva, Switzerland, 2006.
- Mothibe, M.E.; Sibanda, M. African Traditional Medicine: South African Perspective; IntechOpen: London, UK, 2019.
- Tluway, F.; Makani, J. Sickle Cell Disease in Africa: An Overview of the Integrated Approach to Health, Research, Education and Advocacy in Tanzania, 2004–2016. Br. J. Haematol. 2017, 177, 919–929.
- Zang, L.; Baharlooeian, M.; Terasawa, M.; Shimada, Y.; Nishimura, N. Beneficial Effects of Seaweed-Derived Components on Metabolic Syndrome via Gut Microbiota Modulation. Front. Nutr. 2023, 10, 1173225.
- McGann, P.T.; Hernandez, A.G.; Ware, R.E. Sickle Cell Anemia in Sub-Saharan Africa: Advancing the Clinical Paradigm through Partnerships and Research. Blood 2017, 129, 155–161.
- Ameh, S.; Obodozie, O.O.; Afolabi, E.K.; Oyedele, E.O.; Ache, T.; Onanuga, C.; Ibe, M.C.; Inyang, U. Some Basic Requirements for Preparing an Antisickling Herbal Medicine—NIPRISAN®. African J. Pharm. Pharmacol. 2009, 3, 259–264.
- Kitadi, J.M.; Mazasa, P.P.; Tshibangu, D.S.T.; Kasali, F.M.; Tshilanda, D.D.; Ngbolua, K.T.N.; Mpiana, P.T. Ethnopharmacological Survey and Antisickling Activity of Plants Used in the Management of Sickle Cell Disease in Kikwit City, DR Congo. Evid.-Based Complement. Altern. Med. 2020, 2020, 1346493.
- Adejumo, O.E.; Kolapo, A.L.; Folarin, A.O. Moringa oleifera Lam. (Moringaceae) Grown in Nigeria: In Vitro Antisickling Activity on Deoxygenated Erythrocyte Cells. J. Pharm. Bioallied Sci. 2012, 4, 118–122.
- Dafaalla, E.A.A.; Humeida, A.A.K.; Dafaalla, E.A.A.; Humeida, A.A.K. The Effect of Fixed Oil Extracts of Nigella Sativa on Sickle Cells: An in-Vitro Study in Khartoum State -Sudan. World J. Adv. Res. Rev. 2021, 10, 317–321.
- Awor, S.; Bongomin, F.; Kaggwa, M.M.; Pebalo, F.P.; Musoke, D. Prevalence of Use of Herbal Medicines for the Treatment of Sickle Cell Disease in Africa: A Systematic Review and Meta-Analysis. J. Herb. Med. 2023, 42, 100735.
- Mohamed, A.S.; Abd El Dayem, O.Y.; El Shamy, A.M.; El Sakhawy, F.S.; El Gedaily, R.A. Comparative Antisickling and Antioxidant Activities of Pseudobombax Ellipticum Cultivars in Relation to Their Metabolite Profiling Using LC/MS. RSC Adv. 2023, 13, 21327–21335.
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 552535.
- Vamanu, E.; Dinu, L.D.; Pelinescu, D.R.; Gatea, F. Therapeutic Properties of Edible Mushrooms and Herbal Teas in Gut Microbiota Modulation. Microorganisms 2021, 9, 1262.
- del Pozo-Acebo, L.; López de las Hazas, M.C.; Margollés, A.; Dávalos, A.; García-Ruiz, A. Eating MicroRNAs: Pharmacological Opportunities for Cross-Kingdom Regulation and Implications in Host Gene and Gut Microbiota Modulation. Br. J. Pharmacol. 2021, 178, 2218–2245.
- Zhang, H.; Jiang, F.; Zhang, J.; Wang, W.; Li, L.; Yan, J. Modulatory Effects of Polysaccharides from Plants, Marine Algae and Edible Mushrooms on Gut Microbiota and Related Health Benefits: A Review. Int. J. Biol. Macromol. 2022, 204, 169–192.
- Aboderin, F.I.; Oduola, T.; Davison, G.M.; Oguntibeju, O.O. A Review of the Relationship between the Immune Response, Inflammation, Oxidative Stress, and the Pathogenesis of Sickle Cell Anaemia. Biomedicines 2023, 11, 2413.
- Oguntibeju, O. The Interplay among Immune Response, Inflammation, Oxida-Tive Stress and Sickle Cell Anaemia Pathogenesis. J. Forensic Med. 2023, 8, 1–2.
- Fernandes, T.; Garrine, C.; Ferrão, J.; Bell, V.; Varzakas, T. Mushroom Nutrition as Preventative Healthcare in Sub-Saharan Africa. Appl. Sci. 2021, 11, 4221.
- Mshelia Halilu, E. Cultivation and Conservation of African Medicinal Plants for Pharmaceutical Research and Socio-Economic Development; IntechOpen: London, UK, 2022.
- Patel, D.K.; Dutta, S.D.; Ganguly, K.; Cho, S.-J.; Lim, K.-T.; Patel, D.K.; Dutta, S.D.; Ganguly, K.; Cho, S.-J.; Lim, K.-T. Mushroom-Derived Bioactive Molecules as Immunotherapeutic Agents: A Review. Molecules 2021, 26, 1359.
- Łysakowska, P.; Sobota, A.; Wirkijowska, A. Medicinal Mushrooms: Their Bioactive Components, Nutritional Value and Application in Functional Food Production—A Review. Molecules 2023, 28, 5393.
- El-Maradny, Y.A.; El-Fakharany, E.M.; Abu-Serie, M.M.; Hashish, M.H.; Selim, H.S. Lectins Purified from Medicinal and Edible Mushrooms: Insights into Their Antiviral Activity against Pathogenic Viruses. Int. J. Biol. Macromol. 2021, 179, 239–258.
- Das, A.K.; Asif, M.; Hasan, G.M.M.A. A Comparative Study of Fatty Acid Compositions of Three Cultivated Edible Mushroom Species of Bangladesh. J. Agric. Food Res. 2023, 12, 100620.
- Liuzzi, G.M.; Petraglia, T.; Latronico, T.; Crescenzi, A.; Rossano, R. Antioxidant Compounds from Edible Mushrooms as Potential Candidates for Treating Age-Related Neurodegenerative Diseases. Nutrients 2023, 15, 1913.
- Xiao, S.; Wang, Z.; Wang, B.; Hou, B.; Cheng, J.; Bai, T.; Zhang, Y.; Wang, W.; Yan, L.; Zhang, J. Expanding the Application of Tryptophan: Industrial Biomanufacturing of Tryptophan Derivatives. Front. Microbiol. 2023, 14, 1099098.
- Assemie, A.; Abaya, G. The Effect of Edible Mushroom on Health and Their Biochemistry. Int. J. Microbiol. 2022, 2022, 8744788.
- Muszyńska, B.; Grzywacz-Kisielewska, A.; Kała, K.; Gdula-Argasińska, J. Anti-Inflammatory Properties of Edible Mushrooms: A Review. Food Chem. 2018, 243, 373–381.
- Martinez-Medina, G.A.; Chávez-González, M.L.; Verma, D.K.; Prado-Barragán, L.A.; Martínez-Hernández, J.L.; Flores-Gallegos, A.C.; Thakur, M.; Srivastav, P.P.; Aguilar, C.N. Bio-Funcional Components in Mushrooms, a Health Opportunity: Ergothionine and Huitlacohe as Recent Trends. J. Funct. Foods 2021, 77, 104326.
- Cuevas-Cianca, S.I.; Romero-Castillo, C.; Gálvez-Romero, J.L.; Juárez, Z.N.; Hernández, L.R. Antioxidant and Anti-Inflammatory Compounds from Edible Plants with Anti-Cancer Activity and Their Potential Use as Drugs. Molecules 2023, 28, 1488.
- Kozarski, M.; Klaus, A.; Jakovljevic, D.; Todorovic, N.; Vunduk, J.; Petrović, P.; Niksic, M.; Vrvic, M.M.; Van Griensven, L. Antioxidants of Edible Mushrooms. Molecules 2015, 20, 19489–19525.
- Lam-Sidun, D.; Peters, K.M.; Borradaile, N.M. Mushroom-Derived Medicine? Preclinical Studies Suggest Potential Benefits of Ergothioneine for Cardiometabolic Health. Int. J. Mol. Sci. 2021, 22, 3246.
- Cerletti, C.; Esposito, S.; Iacoviello, L. Edible Mushrooms and Beta-Glucans: Impact on Human Health. Nutrients 2021, 13, 2195.
- Timm, T.G.; Costa, T.M.; Alberton, M.D.; Helm, C.V.; Tavares, L.B.B. Mushroom β-Glucans: Application and Innovation for Food Industry and Immunotherapy. Appl. Microbiol. Biotechnol. 2023, 107, 5035–5049.
- Shankar, A.; Sharma, K.K. Fungal Secondary Metabolites in Food and Pharmaceuticals in the Era of Multi-Omics. Appl. Microbiol. Biotechnol. 2022, 106, 3465–3488.
- Nguyen, T.A.N.; Higa, T.; Shiina, A.; Utami, Y.D.; Hiruma, K. Exploring the Roles of Fungal-Derived Secondary Metabolites in Plant-Fungal Interactions. Physiol. Mol. Plant Pathol. 2023, 125, 102021.
- Gupta, S.; Chaturvedi, P.; Kulkarni, M.G.; Van Staden, J. A Critical Review on Exploiting the Pharmaceutical Potential of Plant Endophytic Fungi. Biotechnol. Adv. 2020, 39, 107462.
- Zhao, J.; Hu, Y.; Qian, C.; Hussain, M.; Liu, S.; Zhang, A.; He, R.; Sun, P. The Interaction between Mushroom Polysaccharides and Gut Microbiota and Their Effect on Human Health: A Review. Biology 2023, 12, 122.
- Rani, A.; Saini, K.C.; Bast, F.; Mehariya, S.; Bhatia, S.K.; Lavecchia, R.; Zuorro, A. Microorganisms: A Potential Source of Bioactive Molecules for Antioxidant Applications. Molecules 2021, 26, 1142.
- Hu, W.; Liang, K.; Zhu, H.; Zhao, C.; Hu, H.; Yin, S. Ferroptosis and Its Role in Chronic Diseases. Cells 2022, 11, 2040.
- Tang, H.M.; Cheung, P.C.K. Gallic Acid Triggers Iron-Dependent Cell Death with Apoptotic, Ferroptotic, and Necroptotic Features. Toxins 2019, 11, 492.
- Llabani, E.; Hicklin, R.W.; Lee, H.Y.; Motika, S.E.; Crawford, L.A.; Weerapana, E.; Hergenrother, P.J. Diverse Compounds from Pleuromutilin Lead to a Thioredoxin Inhibitor and Inducer of Ferroptosis. Nat. Chem. 2019, 11, 521–532.
- Wang, Q.; Zennadi, R.; Pantaleo, A.; Secchi, C.; Orecchioni, M. The Role of RBC Oxidative Stress in Sickle Cell Disease: From the Molecular Basis to Pathologic Implications. Antioxidants 2021, 10, 1608.
- El Hajj, S.; Canabady-Rochelle, L.; Gaucher, C. Nature-Inspired Bioactive Compounds: A Promising Approach for Ferroptosis-Linked Human Diseases? Molecules 2023, 28, 2636.
- Ohiri, R.C.; Essien, E.B. Stabilizing Ability and Anti-Sickling Potentials of Ganoderma Lucidum Decoction Extract on Human HbS Erythrocytes Membrane. Am. J. Biomed. Sci 2018, 10, 202–210.
- Yu, T.; Wu, Q.; Liang, B.; Wang, J.; Wu, D.; Shang, X. The Current State and Future Prospects of Auricularia Auricula’s Polysaccharide Processing Technology Portfolio. Molecules 2023, 28, 582.
- Ohiri, R.C.; Odey, O.P. Human Hemoglobin S Erythrocyte-Stabilizing and Antisickling Potential of Extract of Wood Ear Mushroom, Auricularia Auricula (Agaricomycetes). Int. J. Med. Mushrooms 2022, 24, 25–34.
- Anchang, K.Y.; Oko, A.; Richard, S. Toxicological and Positive Changes in Hematological Pattern of Albino Rats Treated with Termitomyces Titanicus: Potential Valorization of an Indigenous Practice in the Treatment of Oris Cancrum (Noma) in Nigeria. J. Pathol. Res. Rev. Rep. 2020, 106, 3.
- Paloi, S.; Kumla, J.; Paloi, B.P.; Srinuanpan, S.; Hoijang, S.; Karunarathna, S.C.; Acharya, K.; Suwannarach, N.; Lumyong, S. Termite Mushrooms (Termitomyces), a Potential Source of Nutrients and Bioactive Compounds Exhibiting Human Health Benefits: A Review. J. Fungi 2023, 9, 112.
- Szućko-Kociuba, I.; Trzeciak-Ryczek, A.; Kupnicka, P.; Chlubek, D. Neurotrophic and Neuroprotective Effects of Hericium Erinaceus. Int. J. Mol. Sci. 2023, 24, 15960.
This entry is offline, you can click here to edit this entry!