Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Hematopoietic stem cells (HSCs) are essential for maintaining overall health by continuously generating blood cells throughout an individual’s lifespan. However, as individuals age, the hematopoietic system undergoes significant functional decline, rendering them more susceptible to age-related diseases. Growing research evidence has highlighted the critical role of epigenetic regulation in this age-associated decline.
- epigenetics
- hematopoietic stem cells
- aging
1. Introduction
Hematopoietic stem cells (HSCs) play a vital role in maintaining a balanced production of all blood cells throughout a lifetime. However, with advancing age, the regenerative capacity and self-renewal potential of HSCs progressively decline while the likelihood of cellular dysfunction significantly rises. This age-related decline in HSC function is accompanied by various molecular and functional changes. One notable change observed in aged HSCs is a compromised self-renewal and differentiation potential, leading to a skewed production of myeloid cells, a decreased output of red blood cells, and a reduced generation of immune cells. These alterations contribute to conditions such as anemia, increased susceptibility to infections, and a higher risk of developing hematopoietic malignancies [1].
In aged HSCs, researchers have identified global epigenetic changes that occur with aging. These changes can either arise randomly through epigenetic drift or result from somatic mutations in genes encoding epigenetic regulatory proteins [2]. Mutations in loci associated with epigenetic modifiers are frequently observed in patients with hematological malignancies, as well as in healthy elderly individuals who are at risk of developing these conditions [3].
2. Epigenetic Regulation of HSCs
2.1. DNA Methylation
DNA methylation, an essential epigenetic modification, is primarily associated with gene repression. It is mediated by a family of DNA methyltransferase enzymes, including DNMT1, DNMT3A, and DNMT3B. DNMT1 plays a crucial role in maintaining pre-existing DNA methylation patterns by recognizing and copying the methylation marks from the parental template strand to the daughter strand [4]. On the other hand, DNMT3A and DNMT3B function as de novo DNA methyltransferases, responsible for establishing new DNA methylation patterns during development and stem cell differentiation [5][6].
DNMT1, an essential DNA methyltransferase, plays a critical role in the self-renewal of HSCs. HSCs lacking DNMT1 exhibit defects in self-renewal capacity, as well as impaired homing to the bone marrow (BM) niche and niche retention. The loss of DNMT1 also exerts specific effects on myeloid progenitor cells, leading to enhanced cell cycling and the inappropriate expression of genes associated with mature lineages [4].
The expression of Dnmt3a is significantly higher in the long-term hematopoietic stem cells (LT-HSCs) compared to progenitor cells and differentiated cells [5]. The loss of Dnmt3a in HSCs impairs differentiation and leads to an expansion of stem cells [5][7].
The Ten-eleven translocation (Tet) methylcytosine dioxygenases are enzymes that catalyze the conversion of DNA 5-methylcytosine (5 mC) to 5-hydroxymethylcytosine (5 hmC), a key step in DNA demethylation. The knockout of Tet2 has been shown to enhance the self-renewal and proliferation of HSCs, disrupting both early and late stages of hematopoiesis, including myeloid and lymphoid differentiation. Additionally, Tet2 knockout has been associated with an increased risk of developing myeloid malignancies [8][9].
Isocitrate dehydrogenase IDH1 (cytosolic protein) and IDH2 (mitochondrial protein) are metabolic enzymes that catalyze the oxidative decarboxylation of isocitrate to produce the α-ketoglutarate (αKG). αKG is essential for the oxidation of 5 mC by TET enzymes. However, IDH1/2 hotspot mutations result in a gain of function in the enzymatic activity, leading to an increased synthesis and accumulation of (R)-2-hydroxyglutarate (2HG) [10].
2.2. Histone Acetylation
Histone acetylation is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs), which play key roles in normal and malignant hematopoiesis.
The MYST family of HATs in humans consists of five members: Tip60, MOZ (KAT6A), MORF (KAT6B), HBO1, and MOF (KAT8) [11]. Among these, KAT6A, KAT6B, and KAT8 play crucial roles in hematopoiesis by catalyzing acetylation on specific lysine residues of histone proteins. KAT6A is responsible for H3K9ac, KAT6B catalyzes H3K23ac/H3K14ac, and KAT8 catalyzes H4K16ac on the regulatory regions of target genes [12]. The activity of these HATs is vital for the generation and development of hematopoietic stem and progenitor cells (HSPCs).
Histone acetyltransferase CREB-binding protein (CBP) and its homolog p300 mediate the deposition of the activating H3K27ac histone marks at gene promoters and enhancers. These transcription co-activators play essential but distinct roles in maintaining normal hematopoiesis. A full dose of CBP, but not p300, is crucial for HSC quiescence and self-renewal [13]. p300, but not CBP, is essential for proper hematopoietic differentiation and proliferation [14].
Histone deacetylases (HDACs) are a class of enzymes that regulate gene expression by deacetylation of lysine residues on histone and nonhistone proteins. In normal hematopoiesis, HDACs are widely involved in the development of various lineages [15]. There are several classes of HDACs, including class I (HDAC1, HDAC2, HDAC3, and HDAC8), class IIa (HDAC4, HDAC5, HDAC7, and HDAC9), class IIb (HDAC6 and HDAC10), class III (sirtuins), and class IV (HDAC11) [16].
HDAC1 and HDAC2 are essential regulators of HSC formation and homeostasis. The ablation of HDAC1 and HDAC2 leads to the depletion of HSCs [15][17]. Additionally, the knockdown of HDAC1 using small interfering RNA promotes myeloid differentiation.
2.3. Histone Methylation
Polycomb repressor complexes 1 and 2 (PRC1 and PRC2) are multi-protein complexes involved in the epigenetic regulation of gene repression. PRC2 is responsible for the trimethylation of histone H3 at lysine 27 (H3K27me3), which is associated with gene silencing. The core components of PRC2 include EZH1/2, EED, and SUZ12. EZH1/2, a histone methyltransferase, catalyzes the addition of methyl groups to H3K27. PRC1 acts downstream of PRC2 and is involved in recognizing the H3K27me3 mark. It further modifies chromatin by adding further modifications, such as monoubiquitination of histone H2A at lysine 119 (H2AK119ub1). PRC1 components include BMI1, RING1A/RING1B, and CBX proteins [18].
Demethylation of H3K27 is regulated by two distinct enzymes: ubiquitously transcribed tetratricopeptide repeat, chromosome X (UTX), also known as KDM6A, and Jumonji-C (JmjC) domain-containing protein-3 (JMJD3), also known as KDM6B. These enzymes specifically recognize and bind to histone mark H3K27me3. Demethylation of H3K27 by KDM6A or JMJD3 results in the conversion of repressive chromatin to a more permissive state. This allows for the recruitment of coactivators and other chromatin-modifying enzymes that promote gene activation [19][20]. KDM6A played an essential role in the hematopoiesis stem cell homing and engraftment [21][22]. The lack of KDM6A is most detrimental to female hematopoiesis due to the partial redundancy with its Y chromosome paralog UTY [23]. The histone demethylase KDM6B is upregulated in a wide range of blood disorders. It is necessary for HSC self-renewal in response to inflammatory and proliferative stress.
H3K4 methyltransferases include MLL (Mixed-Lineage Leukemia) proteins and SET family proteins [24]. MLL1, but not MLL2, is frequently involved in chromosomal rearrangements and the formation of fusion proteins that drive leukemia. Mll1 is required to produce functional HSCs in embryo development [25]. The knockdown of Mll3 or knockout of Mll4 in HSPCs results in impaired differentiation of HSPCs and increased cell expansion [26][27]. The deletion of Setd1a in bone marrow hematopoietic cells blocked B cell differentiation from the pro-B to pre-B cell stage [28].
The H3K4 demethylases KDM5B (Jarid1b) and KDM1A (lysine-specific demethylase 1, LSD1) also play essential roles in the regulation of HSC function. Jarid1b is highly expressed in primitive hematopoietic compartments and is overexpressed in acute myeloid leukemias (AML). The deletion of Jarid1b compromises the HSC self-renewal capacity in mice [29]. On the other hand, LSD1 plays a critical role in repressing HSPC gene expression during hematopoietic differentiation.
H3K9me3 is a histone modification associated with transcriptional repression and the formation of heterochromatin. Several H3K9 methyltransferases, including SUV39H1, SUV39H2, G9a (EHMT2), GLP (EHMT1), and SETDB1, contribute to the establishment of H3K9 methylation marks [30]. During stem cell differentiation, global chromatin rearrangements occur, and the formation of heterochromatin by H3K9 methylation plays a role in regulating HSC differentiation. The inhibition of the histone methyltransferase G9a, which prevents heterochromatin formation, has been shown to delay HSC differentiation [31]. SUV39H1 and SUV39H2 are involved in the maintenance of HSPC functions. The loss of SUV39H function in mice has been shown to disrupt normal hematopoiesis and impair B lymphoid differentiation [32].
2.4. Noncoding RNAs
Non-coding RNAs (ncRNAs) play a crucial role in the regulation of HSC function. These RNA molecules, which do not encode proteins, have been found to participate in diverse cellular processes, including the maintenance, self-renewal, and differentiation of HSCs.
MicroRNAs (miRNAs) are a class of small ncRNAs that regulate gene expression by post-transcriptionally inhibiting translation or by stimulating target mRNA cleavage [33]. In HSCs, specific miRNAs have been identified as key regulators of HSC self-renewal and differentiation, including miR-22, miR-29a, miR-125, miR-126, and the miR-132/122 cluster [33].
Long non-coding RNAs (lncRNAs) have also emerged as important regulators of HSC function. These longer ncRNAs can modulate gene expression at multiple levels, including transcriptional and post-transcriptional regulation. LncRNAs can act as molecular scaffolds, interacting with chromatin-modifying complexes and influencing the epigenetic state of genes involved in HSC regulation [34][35].
2.5. Interplay of Epigenetic Regulators
In general, there is a complex interplay between DNA methylation, histone modifications, and noncoding RNAs in regulating HSC functions. These different layers of epigenetic modifications are often interconnected, with changes in histone modifications being accompanied by alterations in DNA methylation. For example, some of the gene-silencing activities of DNMT3A are intimately tied to chromatin modifications through interactions with specific proteins. DNMT3A can interact with histone modifiers involved in gene repression, such as SUV39H1, SETDB1, and G9A [36][37][38][39], which are linked to H3K9 methylation.
3. Alterations to the Epigenome in HSCs Aging
3.1. Imbalance of Histone Modifications
Aging is accompanied by significant alterations in all layers of epigenetic regulation in HSCs. These shifts are particularly evident in the patterns of histone modifications, which are pivotal for gene expression regulation and chromatin structure organization. The age-related modifications and dysregulation of the epigenetic machinery have profound implications for the functional characteristics of HSCs and can significantly impact the overall process of hematopoiesis during aging.
Sun et al. showed activating H3K4me3 levels increase in old HSCs with broader peaks, especially over genes regulating self-renewal and HSC identity. Additionally, repressive H3K27me3 peaks also broaden with age [40]. The expression of the histone methyltransferase SUV39H1/KMT1A decreases with age in both human and mouse HSCs, resulting in a global reduction in the H3K9me3, disruption of heterochromatin function, and perturbation of B lymphoid differentiation [32][41].
3.2. Age-Related DNA Methylation Changes and Epigenetic Clocks
Age-associated histone modification changes are accompanied by alterations in DNA methylation. Sun et al. found that in aged HSCs, DNA methylation increased at transcription factor binding sites associated with lineage potential and differentiation-promoting genes; at the same time, genes associated with HSC maintenance were relatively hypomethylated [40]. In recent years, these initial observations spurred further advances in the use of DNA methylation as a biomarker to estimate the biological age of various tissues throughout a person’s lifespan. Dubbed “epigenetic clocks,” these innovative tools employ machine learning algorithms that rely on specific CpG sites to establish a connection between the developmental and maintenance processes of biological aging. These clocks provide a means to estimate an individual’s biological age based on epigenetic modifications, contributing to our understanding of the aging process [42].
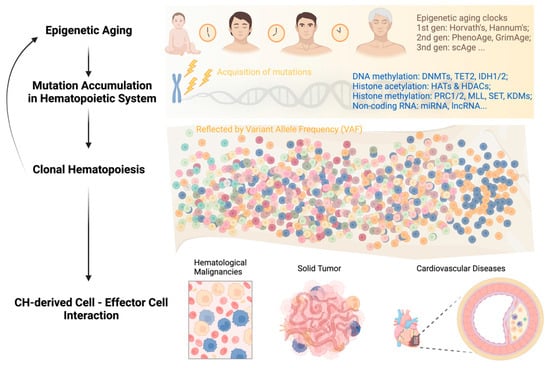
First-generation age estimators, such as Horvath’s clock [43] and Hannum’s clock [44], have found extensive use in aging and cancer research. Horvath’s clock utilizes 353 CpG sites and provides a multi-tissue and cell-type prediction of age. On the other hand, Hannum’s clock focuses on measuring the aging rate using CpG markers from whole blood. The second-generation estimators PhenoAge [45][46] and GrimAge [47] incorporated clinical phenotypes and outcomes into their models, adding further layers of complexity compared to the first-generation clocks.
4. Epigenetics in Acquisition of Clonal Hematopoiesis with Age
Clonal hematopoiesis (CH) is a common phenomenon associated with aging, characterized by the expansion of HSC clones, often defined by the presence of specific mutations, yet without hematologic abnormalities [48][49][50][51][52]. Epigenetic regulators DNMT3A, TET2, and ASXL1 are the three most frequently mutated genes in CH and are also linked to the initiation of myeloid malignancies, including AML [48][52][53][54]. In healthy individuals, normal hematopoiesis is polyclonal, with peripheral blood cells originating from 50,000 to 200,000 HSCs in the bone marrow [55].
While CH itself is asymptomatic, the presence of CH has important health implications in that it is associated with a significantly increased risk of all-cause mortality. In the blood system specifically, CH is associated with an 11-fold increased relative risk of a hematologic malignancy, although the absolute risk is small (5/134 vs. 11/3208 in a median follow-up period of 95 months) and varies depending on the specific gene being affected [56]. The mechanistic roles of DNMT3A and TET2 in malignant hematopoiesis have been extensively summarized elsewhere [57][58]. The impact of CH on health-related outcomes is most notable in non-hematological diseases closely associated with aging, such as cardiovascular disease (CVD) and solid tumors [48][59][60][61][62][63][64][65][66].
While aging leads to CH through the accumulation of mutations, CH can, in turn, accelerate aging. Recent studies have explored the relationship between CH and epigenetic aging by examining several methylation clocks, which have been shown to accurately correlate with chronological age [67][68]. Using whole-genome sequencing and methylation data from 1136 elderly individuals in the Lothian Birth Cohort, it was found that individuals with hematopoietic mutations in various types of CH drivers, including DNMT3A and TET2, had accelerated epigenetic aging as measured by the Horvath clock, a measure of intrinsic age acceleration, and other methylation clocks [68].
In summary, the feedback loop of “aging—genetic mutations—clonal hematopoiesis” can persist throughout a person’s life and impact the survival of both asymptomatic individuals and patients with different types of diseases (Figure 1). Breaking this vicious circle requires multidisciplinary efforts and collaborations among researchers and clinicians in the field. Integrating CH mutation screening into clinical decision-making, including but not limited to hematological malignancies, cardiovascular disease, and solid tumors, can facilitate early diagnosis, risk stratification, and personalized disease management.
Figure 1. Development of clonal hematopoiesis with increasing age at its health implications.
5. Strategies to Alleviate Aging
5.1. Caloric or Dietary Restriction
Caloric or dietary restriction (DR) is known to offer a plethora of health benefits, including enhanced metabolic health, neuroprotection against neurodegenerative diseases, reduced cancer incidence, and extended lifespan through intricate metabolic and epigenetic mechanisms [69][70]. However, the impact of DR on HSCs remains inconclusive. A study by Tao et al. demonstrated that early-onset DR significantly delays the aging process in HSCs, while long-term mid-onset DR improves the regenerative capacity of aging HSCs, particularly in terms of lymphoid outputs [71].
5.2. Small Molecule-Based Therapy
Therapeutic interventions aimed at targeting aging-related epigenetic changes hold promise for preventing or delaying the onset of hematologic disorders. By modulating epigenetic modifiers or rebalancing the cellular epigenetic landscape, it is possible to restore normal epigenetic patterns and rejuvenate aged HSCs. This approach has the potential to enhance HSC function and promote healthy hematopoiesis, opening new avenues for the treatment and prevention of hematologic disorders associated with aging.
CASIN, an inhibitor of Cdc42, has shown potential in rejuvenating old HSCs by targeting both Cdc42 activity and epigenetic reprogramming. Treatment with CASIN ex vivo has been shown to regulate Cdc42 activity and elevate H4K16Ac levels in HSCs, resembling the levels found in young cells. This treatment has demonstrated several beneficial effects, including increasing the percentage of polarized cells, restoring the spatial distribution of H4K16ac, enhancing lymphoid output, and reducing myeloid lineage output [72][73].
5.3. Gene Expression Regulation
Numerous animal studies have highlighted the potential of targeting epigenetic modifiers as effective interventions. One such modifier is SIRT3, a mammalian sirtuin responsible for regulating mitochondrial acetylation. Aged mouse HSCs have been observed to exhibit reduced levels of SIRT3. However, it has been demonstrated that the overexpression of Sirt3 can rescue age-related functional defects in HSCs, ultimately restoring their long-term competitive repopulation ability [74]. Another important epigenetic modifier, SIRT7, has been found to be downregulated in aged murine HSCs. Conversely, the overexpression of Sirt7 has shown promising results whereby it increased the reconstitution capacity of HSCs and reduced the myeloid bias typically associated with aging, thus rescuing myeloid-biased differentiation [75].
5.4. Epigenetic Reprogramming
Epigenetic modifications are pivotal in governing gene expression and maintaining cellular identity. Harnessing the power of induced pluripotent stem cell (iPSC) reprogramming makes it possible to reset or remodel the aberrant epigenetic marks that accumulate during the aging process. This rejuvenation process holds immense promise for enhancing the regenerative potential of HSCs.
This entry is adapted from the peer-reviewed paper 10.3390/epigenomes7040032
References
- De Haan, G.; Lazare, S.S. Aging of hematopoietic stem cells. Blood 2018, 131, 479–487.
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278.
- Buisman, S.C.; de Haan, G. Epigenetic Changes as a Target in Aging Haematopoietic Stem Cells and Age-Related Malignancies. Cells 2019, 8, 868.
- Trowbridge, J.J.; Snow, J.W.; Kim, J.; Orkin, S.H. DNA Methyltransferase 1 Is Essential for and Uniquely Regulates Hematopoietic Stem and Progenitor Cells. Cell Stem Cell 2009, 5, 442–449.
- Challen, G.A.; Sun, D.Q.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.C.; Xi, Y.X.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2012, 44, U23–U43.
- Challen, G.A.; Sun, D.Q.; Mayle, A.; Jeong, M.; Luo, M.; Rodriguez, B.; Mallaney, C.; Celik, H.; Yang, L.B.; Xia, Z.; et al. Dnmt3a and Dnmt3b Have Overlapping and Distinct Functions in Hematopoietic Stem Cells. Cell Stem Cell 2014, 15, 350–364.
- Jeong, M.; Park, H.J.; Celik, H.; Ostrander, E.L.; Reyes, J.M.; Guzman, A.; Rodriguez, B.; Lei, Y.; Lee, Y.J.; Ding, L.; et al. Loss of Dnmt3a Immortalizes Hematopoietic Stem Cells In Vivo. Cell Rep. 2018, 23, 1–10.
- Li, Z.; Cai, X.Q.; Cai, C.L.; Wang, J.P.; Zhang, W.Y.; Petersen, B.E.; Yang, F.C.; Xu, M.J. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518.
- Quivoron, C.; Couronne, L.; Della Valle, V.; Lopez, C.K.; Plo, I.; Wagner-Ballon, O.; Do Cruzeiro, M.; Delhommeau, F.; Arnulf, B.; Stern, M.H.; et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and Is a Recurrent Event during Human Lymphomagenesis. Cancer Cell 2011, 20, 276.
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344.
- Avvakumov, N.; Cote, J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 2007, 26, 5395–5407.
- Khokhar, E.S.; Borikar, S.; Eudy, E.; Stearns, T.; Young, K.; Trowbridge, J.J. Aging-associated decrease in the histone acetyltransferase KAT6B is linked to altered hematopoietic stem cell differentiation. Exp. Hematol. 2020, 82, 43–52.
- Chan, W.I.; Hannah, R.L.; Dawson, M.A.; Pridans, C.; Foster, D.; Joshi, A.; Gottgens, B.; Van Deursen, J.M.; Huntly, B.J.P. The Transcriptional Coactivator Cbp Regulates Self-Renewal and Differentiation in Adult Hematopoietic Stem Cells. Mol. Cell Biol. 2011, 31, 5046–5060.
- Sandberg, M.L.; Sutton, S.E.; Pletcher, M.T.; Wiltshire, T.; Tarantino, L.M.; Hogenesch, J.B.; Cooke, M.P. c-Myb and p300 regulate stem cell proliferation an hematopoietic differentiation. Dev. Cell 2005, 8, 153–166.
- Wang, P.; Wang, Z.; Liu, J. Role of HDACs in normal and malignant hematopoiesis. Mol. Cancer 2020, 19, 5.
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713.
- Heideman, M.R.; Lancini, C.; Proost, N.; Yanover, E.; Jacobs, H.; Dannenberg, J.H. Sin3a-associated Hdac1 and Hdac2 are essential for hematopoietic stem cell homeostasis and contribute differentially to hematopoiesis. Haematologica 2014, 99, 1292–1303.
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349.
- Ding, Y.; Yao, Y.; Gong, X.; Zhuo, Q.; Chen, J.; Tian, M.; Farzaneh, M. JMJD3: A critical epigenetic regulator in stem cell fate. Cell Commun. Signal 2021, 19, 72.
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734.
- Thieme, S.; Gyarfas, T.; Richter, C.; Ozhan, G.; Fu, J.; Alexopoulou, D.; Muders, M.H.; Michalk, I.; Jakob, C.; Dahl, A.; et al. The histone demethylase UTX regulates stem cell migration and hematopoiesis. Blood 2013, 121, 2462–2473.
- Sera, Y.; Nakata, Y.; Ueda, T.; Yamasaki, N.; Koide, S.; Kobayashi, H.; Ikeda, K.; Kobatake, K.; Iwasaki, M.; Oda, H.; et al. UTX maintains the functional integrity of the murine hematopoietic system by globally regulating aging-associated genes. Blood 2021, 137, 908–922.
- Wang, L.; Shilatifard, A. UTX Mutations in Human Cancer. Cancer Cell 2019, 35, 168–176.
- Yang, W.; Ernst, P. Distinct functions of histone H3, lysine 4 methyltransferases in normal and malignant hematopoiesis. Curr. Opin. Hematol. 2017, 24, 322–328.
- Ernst, P.; Fisher, J.K.; Avery, W.; Wade, S.; Foy, D.; Korsmeyer, S.J. Definitive hematopoiesis requires the mixed-lineage leukemia gene. Dev. Cell 2004, 6, 437–443.
- Chen, C.; Liu, Y.; Rappaport, A.R.; Kitzing, T.; Schultz, N.; Zhao, Z.; Shroff, A.S.; Dickins, R.A.; Vakoc, C.R.; Bradner, J.E.; et al. MLL3 Is a Haploinsufficient 7q Tumor Suppressor in Acute Myeloid Leukemia. Cancer Cell 2014, 25, 652–665.
- Santos, M.A.; Faryabi, R.B.; Ergen, A.V.; Day, A.M.; Malhowski, A.; Canela, A.; Onozawa, M.; Lee, J.E.; Callen, E.; Gutierrez-Martinez, P.; et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature 2014, 514, 107–111.
- Tusi, B.K.; Deng, C.W.; Salz, T.; Zeumer, L.; Li, Y.Q.; So, C.W.E.; Morel, L.M.; Qiu, Y.; Huang, S.M. Setd1a regulates progenitor B-cell-to-precursor B-cell development through histone H3 lysine 4 trimethylation and Ig heavy-chain rearrangement. Faseb J. 2015, 29, 1505–1515.
- Stewart, M.H.; Albert, M.; Sroczynska, P.; Cruickshank, V.A.; Guo, Y.P.; Rossi, D.J.; Helin, K.; Enver, T. The histone demethylase Jarid1b is required for hematopoietic stem cell self-renewal in mice. Blood 2015, 125, 2075–2078.
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. 2022, 23, 623–640.
- Ugarte, F.; Sousae, R.; Cinquin, B.; Martin, E.W.; Krietsch, J.; Sanchez, G.; Inman, M.; Tsang, H.; Warr, M.; Passegue, E.; et al. Progressive Chromatin Condensation and H3K9 Methylation Regulate the Differentiation of Embryonic and Hematopoietic Stem Cells. Stem Cell Rep. 2015, 5, 728–740.
- Djeghloul, D.; Kuranda, K.; Kuzniak, I.; Barbieri, D.; Naguibneva, I.; Choisy, C.; Bories, J.C.; Dosquet, C.; Pla, M.; Vanneaux, V.; et al. Age-Associated Decrease of the Histone Methyltransferase SUV39H1 in HSC Perturbs Heterochromatin and B Lymphoid Differentiation. Stem Cell Rep. 2016, 6, 970–984.
- Ortiz, G.G.R.; Mohammadi, Y.; Nazari, A.; Ataeinaeini, M.; Kazemi, P.; Yasamineh, S.; Al-Naqeeb, B.Z.T.; Zaidan, H.K.; Gholizadeh, O. A state-of-the-art review on the MicroRNAs roles in hematopoietic stem cell aging and longevity. Cell Commun. Signal 2023, 21, 85.
- Luo, M.; Jeong, M.; Sun, D.Q.; Park, H.J.; Rodriguez, B.A.T.; Xia, Z.; Yang, L.B.; Zhang, X.T.; Sheng, K.W.; Darlington, G.J.; et al. Long Non-Coding RNAs Control Hematopoietic Stem Cell Function. Cell Stem Cell 2015, 16, 426–438.
- Magilnick, N.; Boldin, M.P. Molecular Moirai: Long Noncoding RNA Mediators of HSC Fate. Curr. Stem Cell Rep. 2018, 4, 158–165.
- Yang, Y.; Liu, R.; Qiu, R.; Zheng, Y.; Huang, W.; Hu, H.; Ji, Q.; He, H.; Shang, Y.; Gong, Y.; et al. CRL4B promotes tumorigenesis by coordinating with SUV39H1/HP1/DNMT3A in DNA methylation-based epigenetic silencing. Oncogene 2015, 34, 104–118.
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 2011, 8, 676–687.
- Epsztejn-Litman, S.; Feldman, N.; Abu-Remaileh, M.; Shufaro, Y.; Gerson, A.; Ueda, J.; Deplus, R.; Fuks, F.; Shinkai, Y.; Cedar, H.; et al. De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat. Struct. Mol. Biol. 2008, 15, 1176–1183.
- Chang, Y.Q.; Sun, L.D.; Kokura, K.; Horton, J.R.; Fukuda, M.; Espejo, A.; Izumi, V.; Koomen, J.M.; Bedford, M.T.; Zhang, X.; et al. MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat. Commun. 2011, 2, 533.
- Sun, D.Q.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging that Reinforce Self-Renewal. Cell Stem Cell 2014, 14, 673–688.
- Keenan, C.R.; Iannarella, N.; Naselli, G.; Bediaga, N.G.; Johanson, T.M.; Harrison, L.C.; Allan, R.S. Extreme disruption of heterochromatin is required for accelerated hematopoietic aging. Blood 2020, 135, 2049–2058.
- Petkovich, D.A.; Podolskiy, D.I.; Lobanov, A.V.; Lee, S.G.; Miller, R.A.; Gladyshev, V.N. Using DNA Methylation Profiling to Evaluate Biological Age and Longevity Interventions. Cell Metab. 2017, 25, 954–960.e6.
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115.
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367.
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging-Us 2018, 10, 573–591.
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384.
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.F.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging-Us 2019, 11, 303–327.
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498.
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752.
- Thompson, D.J.; Genovese, G.; Halvardson, J.; Ulirsch, J.C.; Wright, D.J.; Terao, C.; Davidsson, O.B.; Day, F.R.; Sulem, P.; Jiang, Y.; et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature 2019, 575, 652–657.
- Loh, P.R.; Genovese, G.; Handsaker, R.E.; Finucane, H.K.; Reshef, Y.A.; Palamara, P.F.; Birmann, B.M.; Talkowski, M.E.; Bakhoum, S.F.; McCarroll, S.A.; et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 2018, 559, 350–355.
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487.
- Gao, X.; You, X.; Droin, N.; Banaszak, L.G.; Churpek, J.; Padron, E.; Geissler, K.; Solary, E.; Patnaik, M.M.; Zhang, J. Role of ASXL1 in hematopoiesis and myeloid diseases. Exp. Hematol. 2022, 115, 14–19.
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478.
- Lee-Six, H.; Obro, N.F.; Shepherd, M.S.; Grossmann, S.; Dawson, K.; Belmonte, M.; Osborne, R.J.; Huntly, B.J.P.; Martincorena, I.; Anderson, E.; et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 2018, 561, 473–478.
- Saadatagah, S.; Ballantyne, C.M. Clonal hematopoiesis of indeterminate potential and cardiovascular disease. Transl. Res. 2023, 255, 152–158.
- Venugopal, K.; Feng, Y.; Shabashvili, D.; Guryanova, O.A. Alterations to DNMT3A in Hematologic Malignancies. Cancer Res. 2021, 81, 254–263.
- Bowman, R.L.; Levine, R.L. TET2 in Normal and Malignant Hematopoiesis. Cold Spring Harb. Perspect. Med. 2017, 7, a026518.
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121.
- Natarajan, P. Genomic Aging, Clonal Hematopoiesis, and Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2023, 43, 3–14.
- Vaddavalli, P.L.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in cancer and aging. Trends Genet. 2022, 38, 598–612.
- Lopez-Otin, C.; Pietrocola, F.; Roiz-Valle, D.; Galluzzi, L.; Kroemer, G. Meta-hallmarks of aging and cancer. Cell Metab. 2023, 35, 12–35.
- Lichtman, S.M.; Cohen, H.J.; Muss, H.; Tew, W.P.; Korc-Grodzicki, B. From Assessment to Implementation and Beyond in Cancer and Aging Research. J. Clin. Oncol. 2021, 39, 2217–2225.
- Libby, P.; Sidlow, R.; Lin, A.E.; Gupta, D.; Jones, L.W.; Moslehi, J.; Zeiher, A.; Jaiswal, S.; Schulz, C.; Blankstein, R.; et al. Clonal Hematopoiesis: Crossroads of Aging, Cardiovascular Disease, and Cancer: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 567–577.
- Evans, M.A.; Sano, S.; Walsh, K. Cardiovascular Disease, Aging, and Clonal Hematopoiesis. Annu. Rev. Pathol. 2020, 15, 419–438.
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.; Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 2017, 21, 374–382.e4.
- Robertson, N.A.; Hillary, R.F.; McCartney, D.L.; Terradas-Terradas, M.; Higham, J.; Sproul, D.; Deary, I.J.; Kirschner, K.; Marioni, R.E.; Chandra, T. Age-related clonal haemopoiesis is associated with increased epigenetic age. Curr. Biol. 2019, 29, R786–R787.
- Nachun, D.; Lu, A.T.; Bick, A.G.; Natarajan, P.; Weinstock, J.; Szeto, M.D.; Kathiresan, S.; Abecasis, G.; Taylor, K.D.; Guo, X.; et al. Clonal hematopoiesis associated with epigenetic aging and clinical outcomes. Aging Cell 2021, 20, e13366.
- Green, C.L.; Lamming, D.W.; Fontana, L. Molecular mechanisms of dietary restriction promoting health and longevity. Nat. Rev. Mol. Cell Biol. 2022, 23, 56–73.
- Fontana, L.; Ghezzi, L.; Cross, A.H.; Piccio, L. Effects of dietary restriction on neuroinflammation in neurodegenerative diseases. J. Exp. Med. 2021, 218, e20190086.
- Tao, S.; Wang, Y.T.; Wu, J.Y.; Zeng, T.; Cui, H.; Tao, Z.D.; Lei, L.; Yu, L.; Liu, A.W.; Wang, H.; et al. Long-term mid-onset dietary restriction rejuvenates hematopoietic stem cells and improves regeneration capacity of total bone marrow from aged mice. Aging Cell 2020, 19, e13241.
- Florian, M.C.; Dorr, K.; Niebel, A.; Daria, D.; Schrezenmeier, H.; Rojewski, M.; Filippi, M.D.; Hasenberg, A.; Gunzer, M.; Scharffetter-Kochanek, K.; et al. Cdc42 Activity Regulates Hematopoietic Stem Cell Aging and Rejuvenation. Cell Stem Cell 2012, 10, 520–530.
- Leins, H.; Mulaw, M.; Eiwen, K.; Sakk, V.; Liang, Y.; Denkinger, M.; Geiger, H.; Schirmbeck, R. Aged murine hematopoietic stem cells drive aging-associated immune remodeling. Blood 2018, 132, 565–576.
- Brown, K.; Xie, S.; Qiu, X.; Mohrin, M.; Shin, J.; Liu, Y.; Zhang, D.; Scadden, D.T.; Chen, D. SIRT3 reverses aging-associated degeneration. Cell Rep. 2013, 3, 319–327.
- Mohrin, M.; Shin, J.Y.; Liu, Y.F.; Brown, K.; Luo, H.Z.; Xi, Y.N.; Haynes, C.M.; Chen, D. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 2015, 347, 1374–1377.
This entry is offline, you can click here to edit this entry!