Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Many pathologic conditions are associated with oxidative stress and have increased risk for clinically significant thrombotic events. These conditions include, but are not limited to, disorders of metabolism (e.g., dyslipidemia, diabetes mellitus, and obesity), chronic systemic inflammation, aging, cancer, infection, and blood disorders including hemoglobinopathy, and antiphospholipid syndrome.
- oxidative stress
- cysteine
- CD36
- protein disulfide isomerase
- thrombosis
1. Introduction
Clinically significant thrombotic events promote organ dysfunction, organ failure, and sometimes death [1]. Thrombus formation is a complex, multifaceted process, and oxidative events initiated by reactive species of oxygen, nitrogen, sulfur, and carbon play important roles. These oxidative species induce modifications of cellular constitutions, including lipids and proteins [2]. Oxidative modifications of proteins do not induce clotting on their own, nor are they the final effectors of clotting, but like phosphorylation events that sensitize downstream signaling pathways, oxidative modifications may enhance or limit thrombosis [3] and thus provide additional layers of control of the thrombotic process.
Essential to oxidative post-translational modification of proteins are the reactive species themselves. The chemical reduction or oxidation of molecular oxygen generates reactive oxygen species (ROS), and a detailed review of specific species and their generation mechanisms is provided in a recent review [4]. It is the two-electron oxidants, such as hydrogen peroxide, lipid hydroperoxide, and peroxynitrite, that oxidize thiol side chains of cysteines [5]. Other amino acids are also susceptible to oxidation by ROS; for example, peroxynitrite formed from interaction of superoxide radical and nitric oxide oxidizes tyrosine to form nitrotyrosine [6]. Hypochlorous acid (HOCl), generated by myeloperoxidases in white blood cells, oxidizes methionine and halogenates tyrosine to generate methionine sulfoxide [7] and halo-tyrosine [8], respectively.
2. Cysteine Reactivity: Not All Are Created Equal
Because of its unique redox properties, cysteine plays diverse roles in protein structure and function. Evolutionarily, cysteines are found at both highly conserved and non-conserved sites, and not surprisingly, mutations at these sites often result in pathologic conditions [9]. Cysteine pairs within proteins can form disulfide bonds, thereby contributing importantly to secondary and higher order structure. These residues are often buried from solvent exposure [10]. Cysteines can also be in their free thiol state, where they are exposed to solvent and play non-structural roles, including regulation of enzyme activity (e.g., in kinases) [11][12], metal binding (e.g., Zn-finger transcriptional factors) [13], catalytic redox reactions [14][15], and catalytic nucleophilic reactions (e.g., caspases and phosphatases) [16]. The nucleophilicity of the thiol is determined by the stabilization of the negatively charged thiolate [17], a physicochemical property of the sulfur atom.
Deprotonation of the cysteine thiol is a critical step in generating the nucleophilic thiolate and depends on the acid dissociation constant or pKa of the cysteine and the local pH of the environment [18]. If pKa is lower than the pH, the fraction of cysteine in the deprotonated form is higher. At physiologic pH ~7.4, the cysteines with pKa of less than 7.4 should have a higher fraction in the thiolate form and are thus more “reactive”. However, it should be noted that the pKa of the thiol group of free cysteine is ~8.3 and can range substantially (from 3.5 to 13 depending on the microenvironment) [19][20]. The pH of the local environment is also not uniform [20]; lysosomes, for example, have much lower pH (pH 4–5) relative to the cytosol (pH 6.8–7.4) and mitochondria (pH~8.0 within mitochondrial matrix) [21]. As the pKa is linked to the pH of the aqueous environment, solvent accessibility (e.g., buried vs. exposed) influences cysteine reactivity. Additional factors that influence thiolate formation include proximal structural elements of the protein that can stabilize the negative charge [22]. Hydrogen bond donors, such as the hydroxyl group of threonines as in thioredoxins [23], have a partial positive charge from the hydrogen allowing for stability in the thiolate through bonding with the sulfur atom [19]. Electropositive (basic) amino acids including arginine and histidine afford additional ionic interaction with the negative charge of the sulfur and stabilize the thiolate for redox events [24]. Similarly, positively charged macrodipoles of alpha helices afford electrostatic interactions with the thiolate for stability [25].
Cysteine reactivity is also controlled by the redox potential of the thiol. Electrons are poised to proceed from the more electronegative to the more electropositive redox potential, with the potential ranging substantially in biologic systems (from −480 to −80 mV) [26]. In an isolated system, these potentials could be calculated for a thiol using different pairs of redox couplers (e.g., reduced glutathione (GSH) with oxidized glutathione (GSSG); reduced dithiothreitol with oxidized dithiothreitol; cysteines and cystines); however, the dynamics of redox potential within a biological system are quite complex and difficult to calculate owing to the different redox couplers in any given location [20]. This suggests that cysteines could be more reactive within different sub-compartments of the cell, with a classic example being the endoplasmic reticulum where the environment is more oxidizing to facilitate oxidative protein folding.
Lastly, an environmental factor that influences cysteine reactivity is proximity to the source of oxidants. Many sources within a cell promote electron leakages (and transfers) that could generate reactive oxygen species [27]. A protein that is closer to a source of oxidant is more likely to encounter the reactive species prior to the oxidant degrading through an efficient antioxidative mechanism or through competing oxidizing targets. Proteins further away from a source of oxidant are protected as the oxidants have less of a chance to reach the target. Evidence connecting proximity of a protein to an oxidant source and oxidation of that protein were demonstrated in studies of an epidermal carcinoma cell line [28]. In these cells, Epidermal Growth Factor (EGF) binding to its receptor (EGFR) [28][29] promotes receptor dimerization and activation, allowing for downstream signaling for proliferation, differentiation, growth, and survival [30]. The proximity of EGFR to NADPH oxidase was shown to promote cysteine oxidation of the receptor, enhancing kinase activity and downstream signaling.
3. Cysteine Oxoforms
Oxidation of the cysteine thiolate generates many sulfur oxoforms. A depiction of these oxoforms is presented in Figure 1. The sulfur atom attains valences between −2 and +6 oxidation states [5]. Oxidation of the thiolate by peroxide generates a sulfenic acid, which is an intermediate oxoform that is labile and susceptible to convert to other oxoforms [5]. Further oxidation of the sulfenic acid could generate sulfinic and sulfonic acid. Oxidation of the sulfinic acid is reversible with sulfinic acid reductase [31], whereas the sulfonic acid is currently believed to be irreversible. Sulfenic acids could also lead to the formation of other oxidative cysteine modifications, including cysteine glutathionylation (by reaction with glutathione), sulfenamides (reacting with an amine) [32], thiolsulfenates (reacting with a nearby sulfenic acid) [5], and disulfides [5][33]. In some instances, other cysteine oxoforms could be converted over to a sulfenic acid (as is the case for nitrosothiols) [5]. These oxoforms emphasize the significance of the sulfenic acid as a “gateway” modification that may be able to be targeted pharmacologically.
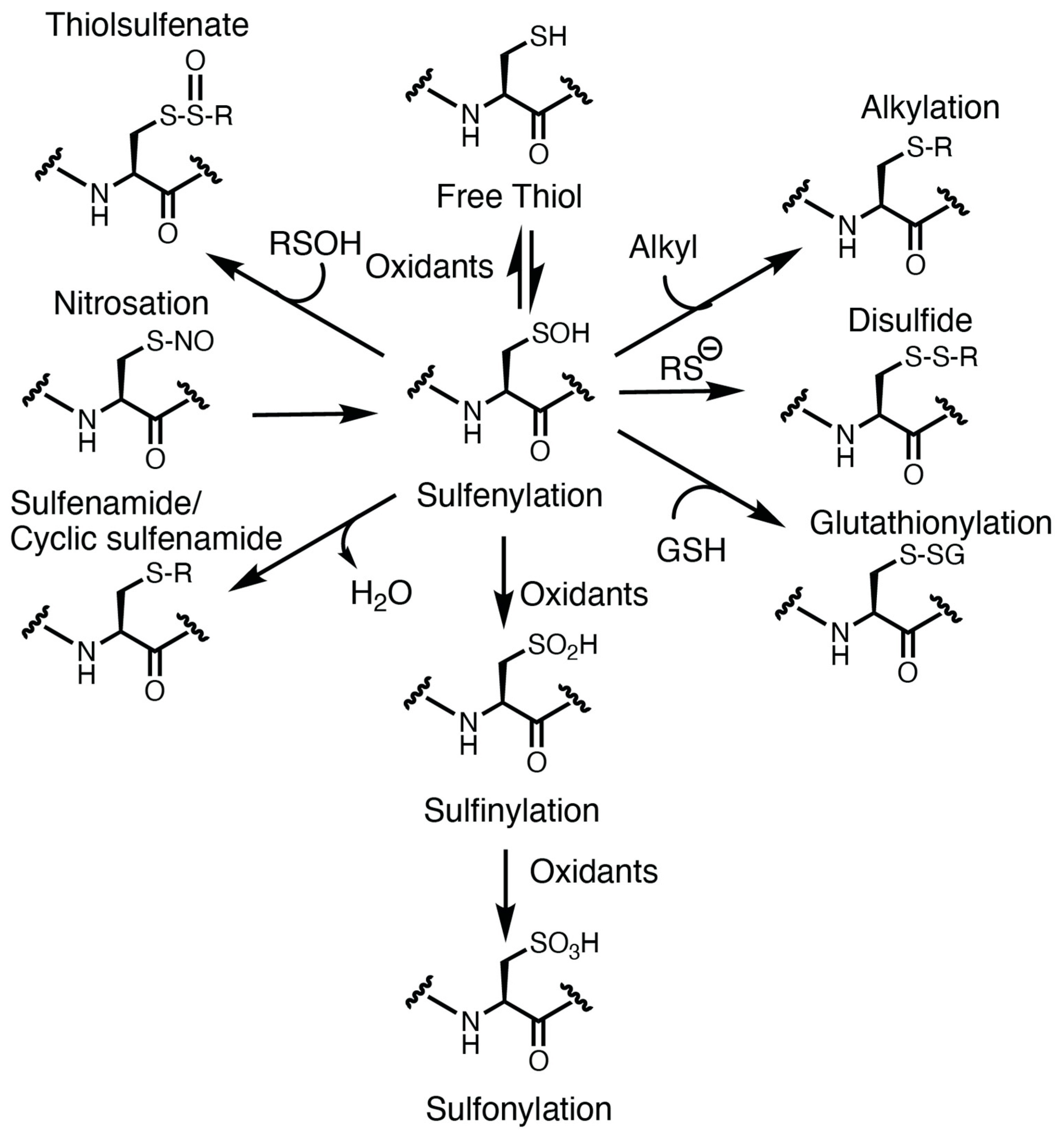
Figure 1. Oxidative modification of cysteine. The cysteine thiolate is nucleophilic and susceptible to oxidation. Oxidation of the thiol generates a transient and labile modification: the sulfenic acid (sulfenylation). Sulfur oxidation by nitric oxide or nitrosothiols could also lead to sulfenic acid formation. Sulfenylation is a cysteine oxoform that is at the crossroad of further oxidative cysteine modification, including sulfinylation, sulfonylation, glutathionylation, disulfide, alkylation, thiolsulfenate, and sulfenamides.
This entry is adapted from the peer-reviewed paper 10.3390/antiox13010083
References
- Wendelboe, A.M.; Raskob, G.E. Global Burden of Thrombosis. Circ. Res. 2016, 118, 1340–1347.
- Demasi, M.; Augusto, O.; Bechara, E.J.; Bicev, R.N.; Cerqueira, F.M.; da Cunha, F.M.; Denicola, A.; Gomes, F.; Miyamoto, S.; Netto, L.E.; et al. Oxidative Modification of Proteins: From Damage to Catalysis, Signaling, and Beyond. Antioxid. Redox Signal. 2021, 35, 1016–1080.
- Yang, M.; Smith, B.C. Cysteine and methionine oxidation in thrombotic disorders. Curr. Opin. Chem. Biol. 2023, 76, 102350.
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515.
- Gupta, V.; Carroll, K.S. Sulfenic acid chemistry, detection and cellular lifetime. Biochim. Biophys. Acta (BBA) Gen. Subj. 2014, 1840, 847–875.
- Yoon, S.; Eom, G.H.; Kang, G. Nitrosative Stress and Human Disease: Therapeutic Potential of Denitrosylation. Int. J. Mol. Sci. 2021, 22, 9794.
- Aussel, L.; Ezraty, B. Methionine Redox Homeostasis in Protein Quality Control. Front. Mol. Biosci. 2021, 8, 665492.
- Sun, H.; Jia, H.; Kendall, O.; Dragelj, J.; Kubyshkin, V.; Baumann, T.; Mroginski, M.-A.; Schwille, P.; Budisa, N. Halogenation of tyrosine perturbs large-scale protein self-organization. Nat. Commun. 2022, 13, 4843.
- Marino, S.M.; Gladyshev, V.N. Cysteine Function Governs Its Conservation and Degeneration and Restricts Its Utilization on Protein Surfaces. J. Mol. Biol. 2010, 404, 902–916.
- Chiu, J.; Hogg, P.J. Allosteric disulfides: Sophisticated molecular structures enabling flexible protein regulation. J. Biol. Chem. 2019, 294, 2949–5908.
- Awoonor-Williams, E.; Rowley, C.N. How Reactive are Druggable Cysteines in Protein Kinases? J. Chem. Inf. Model. 2018, 58, 1935–1946.
- Liu, R.; Zhan, S.; Che, Y.; Shen, J. Reactivities of the Front Pocket N-Terminal Cap Cysteines in Human Kinases. J. Med. Chem. 2022, 65, 1525–1535.
- Pace, N.J.; Weerapana, E. Zinc-Binding Cysteines: Diverse Functions and Structural Motifs. Biomolecules 2014, 4, 419–434.
- Yang, M.; Flaumenhaft, R. Oxidative Cysteine Modification of Thiol Isomerases in Thrombotic Disease: A Hypothesis. Antioxid. Redox Signal. 2021, 35, 1134–1155.
- Gaspar, R.S.; Gibbins, J.M. Thiol Isomerases Orchestrate Thrombosis and Hemostasis. Antioxid. Redox Signal. 2021, 35, 1116–1133.
- Ramos-Guzmán, C.A.; Ruiz-Pernía, J.J.; Zinovjev, K.; Tuñón, I. Unveiling the Mechanistic Singularities of Caspases: A Computational Analysis of the Reaction Mechanism in Human Caspase-1. ACS Catal. 2023, 13, 4348–4361.
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679.
- Marino, S.M.; Gladyshev, V.N. Analysis and Functional Prediction of Reactive Cysteine Residues. J. Biol. Chem. 2012, 287, 4419–4425.
- Roos, G.; Foloppe, N.; Messens, J. Understanding the pKa of Redox Cysteines: The Key Role of Hydrogen Bonding. Antioxid. Redox Signal. 2013, 18, 94–127.
- Bak, D.W.; Bechtel, T.J.; Falco, J.A.; Weerapana, E. Cysteine reactivity across the subcellular universe. Curr. Opin. Chem. Biol. 2019, 48, 96–105.
- Yang, R.; Zhu, T.; Xu, J.; Zhao, Y.; Kuang, Y.; Sun, M.; Chen, Y.; He, W.; Wang, Z.; Jiang, T.; et al. Organic Fluorescent Probes for Monitoring Micro-Environments in Living Cells and Tissues. Molecules 2023, 28, 3455.
- Mazmanian, K.; Chen, T.; Sargsyan, K.; Lim, C. From quantum-derived principles underlying cysteine reactivity to combating the COVID-19 pandemic. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1607.
- Røhr, K.; Hammerstad, M.; Andersson, K.K. Tuning of Thioredoxin Redox Properties by Intramolecular Hydrogen Bonds. PLoS ONE 2013, 8, e69411.
- Karala, A.-R.; Lappi, A.-K.; Ruddock, L.W. Modulation of an Active-Site Cysteine pKa Allows PDI to Act as a Catalyst of both Disulfide Bond Formation and Isomerization. J. Mol. Biol. 2010, 396, 883–892.
- Kortemme, T.; Creighton, T.E. Ionisation of Cysteine Residues at the Termini of Model α-Helical Peptides. Relevance to Unusual Thiol pKaValues in Proteins of the Thioredoxin Family. J. Mol. Biol. 1995, 253, 799–812.
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157.
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383.
- Paulsen, C.E.; Truong, T.H.; Garcia, F.J.; Homann, A.; Gupta, V.; Leonard, S.E.; Carroll, K.S. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 2011, 8, 57–64.
- Truong, T.H.; Ung, P.M.-U.; Palde, P.B.; Paulsen, C.E.; Schlessinger, A.; Carroll, K.S. Molecular Basis for Redox Activation of Epidermal Growth Factor Receptor Kinase. Cell Chem. Biol. 2016, 23, 837–848.
- Rosenkranz, A.A.; Slastnikova, T.A. Epidermal Growth Factor Receptor: Key to Selective Intracellular Delivery. Biochemistry 2020, 85, 967–1092.
- Akter, S.; Fu, L.; Jung, Y.; Conte, M.L.; Lawson, J.R.; Lowther, W.T.; Sun, R.; Liu, K.; Yang, J.; Carroll, K.S. Chemical proteomics reveals new targets of cysteine sulfinic acid reductase. Nat. Chem. Biol. 2018, 14, 995–1004.
- Petroff, J.T.; Omlid, S.M.; Haloi, N.; Sith, L.; Johnson, S.; McCulla, R.D. Reactions of sulfenic acids with amines, thiols, and thiolates studied by quantum chemical calculations. Comput. Theor. Chem. 2020, 1189, 112979.
- Rehder, D.S.; Borges, C.R. Cysteine sulfenic Acid as an Intermediate in Disulfide Bond Formation and Nonenzymatic Protein Folding. Biochemistry 2010, 49, 7748–7755.
This entry is offline, you can click here to edit this entry!