2. “Starting from Scratch”—Targeting the Energy Substrate Selection Stage
In chronic renal failure, the primary pathophysiological mechanism at the energy substrate selection stage is the disorder in fatty acid utilization, and restoring normal fatty acid oxidation presents a potential therapy for this condition [
19].
L-carnitine promotes the mitochondrial matrix transport of long-chain fatty acids, regulates fatty acid β-oxidation, and possesses antioxidant and free radical scavenging properties. It has exhibited benefits for various diseases characterized by low carnitine levels or impaired fatty acid oxidation [
131,
132,
133]. In the human body, L-carnitine mainly relies on dietary intake and endogenous synthesis in the liver and kidneys. The balance of carnitine homeostasis is maintained through glomerular filtration and tubular reabsorption in the kidneys [
134]. Chronic renal failure patients often suffer from carnitine deficiency due to dietary restrictions, renal dysfunction, and continuous loss during dialysis. Thus, oral or intravenous supplementation of L-carnitine presents a potential therapy for correcting the abnormal metabolic reprogramming in these patients [
134,
135]. Several clinical randomized controlled trials of L-carnitine supplementation have been conducted in patients undergoing hemodialysis or peritoneal dialysis for chronic renal failure; however, the existing results are somewhat controversial [
136,
137,
138,
139,
140].
PGC-1α is a key transcription factor that regulates mitochondrial biogenesis and function, as well as a crucial upstream regulator of fatty acid oxidation [
141,
142]. Experimental models of chronic kidney disease in mice and patients with chronic renal failure exhibited low levels of PGC-1α expression. Pharmacological activation of PGC-1α presented a potential therapy for improving energy metabolism in patients with chronic renal failure [
143,
144,
145,
146,
147]. A novel selective PGC-1α small-molecule agonist, ZLN005, has been validated in mice as promoting fatty acid oxidation and mitochondrial biogenesis. It was shown to improve insulin resistance and ketone tolerance in diabetic mouse models and to alleviate fibrosis and lipid accumulation in a unilateral ureteral obstruction (UUO) mouse model [
148,
149]. The traditional Chinese medicine prescription Shen Shuai II stimulated PGC-1α expression and improved mitochondrial functional protein expression and energy production in hypoxia-treated renal tubular epithelial cells (HK-2) and a 5/6 nephrectomy rat model of CKD. It also inhibited hypoxia-induced fibrosis in HK-2 cells [
150]. Unfortunately, this treatment prescription lacks data related to fatty acid metabolism. The plant extract sulforaphane enhanced the expression of PGC-1α and nuclear respiratory factor 1 (NRF1) by suppressing the fatty acid intake membrane receptor CD36 and enhancing the expression of the key fatty acid oxidation enzyme CTP1A, reducing lipid deposition in a UUO rat model. It also improved the tricarboxylic acid cycle by increasing the expression and activity of mitochondrial functional proteins [
151]. Furthermore, the plant extract huperzine A glucoside has been shown to activate PGC-1α transcription, but its specific pharmacological effects require further investigation [
152].
The tissue expression of PPARα positively correlates with mitochondrial density and fatty acid β-oxidation levels, thus playing an important role in lipid metabolism [
153]. Patients with chronic kidney failure exhibited reduced expression of renal PPARα. Mouse models further corroborated the link between low PPARα expression and the progression of fibrosis, suggesting that PPARα agonists hold potential as therapeutic drugs for chronic kidney failure [
20,
154,
155]. Fibrates, the most commonly used PPARα agonists, are mainly excreted by the kidneys, thus limiting their use in patients with chronic kidney failure due to potential kidney-related complications. The novel fibrate pemafibrate, mainly excreted by bile, regulated fatty acid metabolism by activating renal PPARα and its target genes, leading to the inhibition of kidney fibrosis and the expression of inflammatory markers in UUO mice. Additionally, it improved plasma creatinine and blood urea nitrogen levels, as well as kidney fibrosis in CKD mouse models, consequently reducing renal inflammation and oxidative stress levels [
156].
Cpt1A is a crucial rate-limiting enzyme in fatty acid metabolism. Reduced expression of Cpt1A in patients with chronic kidney failure is associated with fibrosis. Overexpression of Cpt1A in a mouse model of CKD restored fatty acid metabolism in the fibrotic kidney, which improved mitochondrial homeostasis and consequently ameliorated both renal fibrosis and kidney function [
157]. Cpt1A agonists are potential drugs for targeting the energy substrate selection stage to improve fibrosis in chronic kidney failure. Resveratrol and its derivative, BEC2, have been experimentally confirmed as directly activating Cpt1A, thus accelerating long-chain fatty acid β-oxidation, but this class of drugs has not yet been used in animal CKD models [
158,
159].
3. Strive for “Precision Strike”—Targeting the Mitochondrial Respiratory Chain
Mitochondrial damage and dysfunction represent the primary pathogenic events in chronic kidney failure, with the dysfunction of the respiratory chain serving as the central component. Restoring the function of the mitochondrial respiratory chain is crucial in preventing the progression of chronic kidney failure [
160].
Coenzyme Q10 (CoQ10) serves as both an electron carrier in the respiratory chain and an effective scavenger of reactive oxygen species [
161]. The reduction in CoQ10 in the plasma of chronic kidney failure patients results in the diminished efficiency of electron transport in the respiratory chain, alterations in mitochondrial membrane potential, escalated production of reactive oxygen species, and a cascade of pathological changes [
162]. As a result, the supplementation of CoQ10 not only enhances electron transport efficiency in the respiratory chain to facilitate ATP production, but also ameliorates abnormal fatty acid metabolism in diabetic and obese mice and patients with chronic kidney failure through the upregulation of PGC-1α expression. Additionally, it inhibits the depolarization of the mitochondrial membrane potential, thereby reducing oxidative stress markers in chronic kidney failure patients [
162,
163,
164,
165,
166]. Moreover, CoQ10 supplementation demonstrated the ability to decrease proteinuria in a rat model of subtotal nephrectomy chronic kidney disease and in patients with primary CoQ10-induced kidney failure, consequently contributing to an improvement in kidney function to some extent. A large dosage of oral CoQ10 supplementation can successfully eliminate proteinuria and preserve normal kidney function in children with inherited mutations of
CoQ2,
CoQ6, and
CoQ8b genes [
90,
123,
167,
168].
RP81-MNP is a nanocapsule-encapsulated renal enzyme stimulant that targets the proximal tubules of the kidney. RP81-MNP administration mainly upregulated the expression of mitochondrial respiratory chain complex I Nd1, Nd3–5 subunits and enhanced the reduction state of complex I to reduce cisplatin-induced renal tubular damage and excessive ROS production in a mouse model of CKD [
169]. GC4419, a novel small molecule superoxide dismutase (SOD) mimic, demonstrated the ability to reduce excessive superoxide anion production induced by cisplatin. This was achieved by inhibiting the abnormal activity of mitochondrial respiratory chain complex I, leading to improvements in renal tubule necrosis, interstitial fibrosis, and the protection of kidney function in a mouse model of CKD [
170].
Mitochondrial acid MA-5 is a newly synthesized indole derivative, which can regulate mitochondrial ATP synthesis and clear mitochondrial ROS production by promoting ATP synthase oligomerization and forming a supercomplex with mitofilin/Mic60 to improve mitochondrial dysfunction. Its nephroprotective effect has been further demonstrated in oxidative stress cell models and cisplatin-induced mouse nephropathy models [
171,
172,
173]. The emergence of MA-5 provides a new strategy for mitochondrial-targeted therapy for chronic renal failure.
Mitochondrial complex I and cytochrome c are considered to be the targets of flavonoids [
174]. Pre-administration of curcumin effectively mitigated the decline in respiratory chain complex I and V activities in a rat 5/6 nephrectomy model. This protective effect on the respiratory chain complex ameliorated excessive ROS production and renal structural damage. Unfortunately, the study on the efficacy of this drug has been limited to preventive administration [
175,
176,
177]. Quercetin stimulated mitochondrial biogenesis and suppressed the production of reactive oxygen species by elevating the concentration of the electron carrier cytochrome c and inhibiting the generation of superoxide anions by mitochondrial complex I. Consequently, it suppressed inflammation and the expression of apoptosis factors in the rat UUO model [
174,
178,
179]. Meanwhile, the mixed preparation of curcumin and quercetin, Oxy-Q, was confirmed in a phase I clinical trial to improve early graft function in deceased donor kidney transplant recipients. Further promotion of flavonoid preparations in the treatment of chronic kidney failure is anticipated [
180].
The renal protective effect of non-flavonoid polyphenols, resveratrol, has been verified in various models of acute kidney injury [
181,
182]. Resveratrol could also improve mitochondrial ATP synthesis in the kidneys and reverse depolarization of mitochondrial membranes to alleviate glomerular injury in the 5/6 nephrectomy CKD rat model by increasing the expression of ATP synthase subunit beta and cytochrome c oxidase subunit I protein, and by exposing mesangial cells to TGF-β1 [
183]. Unfortunately, the poor bioavailability of resveratrol has limited the translation of animal experiments to clinical trials. Improving the delivery of the drug, such as nanoencapsulation, is critical for its further clinical promotion [
184].
Extracts of the traditional Chinese medicine formula Zhen Wu Decoction enhanced the expression of representative subunits NDUFB8, SDHB, UQCRC2, COX-I, and ATP5A of mitochondrial respiratory complexes I-V in the kidneys of mice in a UUO model, restoring oxidative phosphorylation and improving kidney fibrosis and renal function damage [
185]. However, since this research was based on a composite formula, the specific effective ingredients need further clarification.
Preventive administration of the member of the vitamin E family, γ-tocotrienol, could effectively prevent the decrease in the activity of complexes I, III, and F
0F
1-ATPase after ischemia/reperfusion injury, preserve ATP levels in the renal cortex, and alleviate renal tubular injury and the post-injury inflammatory response [
186]. However, as with curcumin, this drug is still in the stage of preventive administration and lacks verification in CKD models. It is unknown whether it has the same renal protective effect on patients with chronic kidney failure.
4. “Stepping on the Brake”—Targeting Mitochondrial Oxidative Stress
The causal relationship between oxidative stress and respiratory chain dysfunction is a primary contributor to the development and progression of chronic kidney failure. Addressing oxidative stress, particularly that originating from mitochondria, holds promise as a therapeutic approach for managing or ameliorating chronic kidney failure [
74,
76]. Traditional drugs that primarily act on energy substrate selection and mitochondrial respiratory chain stages can improve mitochondrial function to a greater or lesser extent while also having some degree of free radical scavenging effects. The primary hindrance to the antioxidant effect is the low concentrations of drugs in the mitochondria. The emergence of novel antioxidants specifically targeted at the mitochondria has facilitated the targeting of mitochondrial oxidative stress [
12].
Mito molecules, such as MitoQ and Mito-TEMPO, are mitochondrial-targeted antioxidants traditionally linked to triphenylphosphonium (TPP) cations. Their specific delivery primarily relies on the electrostatic attraction between the outer TPP carrying a positive charge and the high transmembrane potential of the mitochondria [
187]. MitoQ is formed by covalently connecting the quinone part to TPP. Upon entry into the mitochondria, the quinone part integrated into the mitochondrial lipid bilayer and underwent reduction by the respiratory chain, forming a quinol derivative. This derivative acted as a potent antioxidant, preventing lipid peroxidation and restoring activity through the respiratory chain cycle [
188]. Currently, MitoQ has been validated to delay age-related kidney fibrosis in a mouse aging model and improve vascular dysfunction in patients with chronic kidney failure, suggesting its potential for application in chronic kidney failure patients [
189,
190]. Mito-TEMPO, an SOD mimic composed of peroxynitrite and TPP coupling, effectively reversed DNA methylation and reduced kidney fibrotic changes in an NDRG2-dependent manner, leading to a notable enhancement in renal function in a rat model of chronic kidney failure [
191,
192]. Furthermore, mitochondrial-targeted quinone analogs such as SkQ1 and SkQR1, as well as the SOD mimic Mito-CP, have demonstrated renal protective effects in various acute kidney injury models, though their verification in chronic kidney failure models is still pending [
193,
194].
Sodium tanshinone IIA sulfonate (SS) peptides are a class of cell-penetrating peptides with a specific mitochondrial-targeting sequence. They eliminate oxygen free radicals through tyrosine or dimethyltyrosine residues and are currently considered highly promising mitochondrial-targeted efficient antioxidants [
195]. SS-31 penetrates the mitochondria in a manner independent of membrane potential and accumulates in the inner mitochondrial membrane to eliminate reactive oxygen species, thereby inhibiting the opening of mitochondrial permeability transition pores and the release of cytochrome c [
196]. Administration of SS-31 effectively improved glomerulosclerosis and tubulointerstitial fibrosis in a rat 5/6 nephrectomy and unilateral ureteral obstruction (UUO) model, reduced renal function damage and proteinuria, and effectively prevented the transition from acute ischemic AKI to CKD [
197,
198,
199,
200]. SS-20, another SS peptide targeting the inner mitochondrial membrane, shares the same antioxidant mechanism as SS-31 but is not as widely utilized. While clearing mitochondrial reactive oxygen species, it effectively improved mitochondrial respiratory chain efficiency and ameliorated renal dysfunction and inflammation progression in a mouse model of chronic kidney failure [
201]. Recently, electrostatically complexed SS-31 nanopolymer chains formed using anionic hyaluronic acid and cationic chitosan have achieved a breakthrough in targeting acute kidney injury after systemic administration, providing insights for targeting chronic kidney injury [
202]. Additionally, mtCPP-1, a mitochondria-targeting peptide designed based on the structure of SS-31, has shown better mitochondrial-targeting ability than SS-31 [
203]. Therefore, before clinical application in chronic kidney failure patients, the focus of drug improvement for SS-31 should be on improving the targeting of chronic kidney injury and mitochondrial targeting.
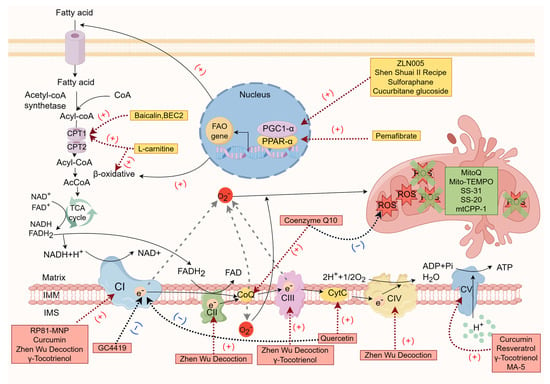
The potential therapeutic agents for chronic renal failure targeting the mitochondrial respiratory chain are shown in Table 1, and the mechanisms of action of the potential therapeutic drugs for chronic renal failure are shown in Figure 1.
Figure 1. The mechanisms of action of potential therapeutic drugs for chronic renal failure (by Figdraw).
Table 1. Potential therapeutic agents for chronic renal failure targeting the mitochondrial respiratory chain.
| Drug Name |
The Main Action Stage |
Mechanism |
Current Usage Status |
| L-carnitine [131,132,133,134,135,136,137,138,139,140,204] |
Energy substrate selection |
Mediates fatty acid transport and promotes the tricarboxylic acid cycle |
Validated by randomized clinical trials in patients with hemodialysis and peritoneal dialysis with chronic renal failure, but the results were controversial |
| ZLN005 [148,149] |
Energy substrate selection |
PGC-1α agonists, promotes fatty acid oxidation, mitochondrial biogenesis and function |
Phenotypic improvement validation of mouse model of diabetes mellitus and UUO |
| Shen Shuai Ⅱ recipe [150] |
Energy substrate selection |
Activates PGC-1α and regulates mitochondrial dynamics |
Phenotypic improvement validation of rat 5/6 nephrectomy CKD model |
| Sulforaphane [151] |
Energy substrate selection |
Enhances PGC-1α and NRF1 expression, improves lipid metabolism and mitochondrial biogenesis |
Phenotypic improvement validation of rat UUO model |
| Cucurbitane glucoside [152] |
Energy substrate selection |
Activates PGC-1α |
Lack of animal model validation |
| Pemafibrate [156] |
Energy substrate selection |
PPARα agonist, regulates fatty acid metabolism |
Phenotypic improvement validation of mouse UUO and purine-induced CKD models |
| Baicalin, BEC2 [158,159] |
Energy substrate selection |
CPT1A agonist, accelerates β oxidation of long-chain fatty acids |
Not verified by mouse CKD model |
| Coenzyme Q10 [90,123,162,163,164,165,166,167,168] |
Mitochondrial respiratory chain |
improves the electron transport efficiency of the respiratory chain, activates PGC-1α to improve fatty acid metabolism, and inhibits mitochondrial membrane potential depolarization |
Phenotypic improvement validation of a rat renal hemirectomy CKD model and patients with chronic renal failure, large-scale
clinical randomized controlled trials were lacking |
| RP81-MNP [169] |
Mitochondrial respiratory chain |
Upregulates the expression of mitochondrial complex I subunit and enhances the reduction state of complex I |
Phenotypic improvement validation of cisplatin-induced mouse CKD model |
| GC4419 [170] |
Mitochondrial respiratory chain |
Inhibits mitochondrial complex I aberrant activity |
Phenotypic improvement validation of cisplatin-induced mouse CKD model |
| MA-5 [171,172,173] |
Mitochondrial respiratory chain |
Promotes ATP synthase oligomerization and forms a supercomplex with mitofilin/Mic60 |
Phenotypic improvement validation of cisplatin-induced mouse nephropathy model |
| Curcumin [176,177] |
Mitochondrial respiratory chain |
Maintains complexes I, V activity |
Prophylactic administration was used to verify the protective effect of renal function in rat 5/6 nephrectomy CKD model |
| Quercetin [174,178,179] |
Mitochondrial respiratory chain |
Enhances cytochrome C concentration and inhibits the generation of superoxide anion by complex I |
Validation of phenotypic improvement in rat UUO model |
| Resveratrol [183] |
Mitochondrial respiratory chain |
Increases the expression of ATP synthase β and cytochrome c oxidase subunit I protein, promotes ATP synthesis, and reverses mitochondrial hyperpolarization membrane potential |
Validation of phenotypic improvement in rat 5/6 nephrectomy CKD model |
| ZhenWu Decoction [185] |
Mitochondrial respiratory chain |
Enhances mitochondrial respiratory complex I-V subunit expression to restore oxidative phosphorylation |
Validation of phenotypic improvement in rat UUO model |
| γ-Tocotrienol [186] |
Mitochondrial respiratory chain |
Maintains complex I, III and F0F1-ATPase activity |
Prophylactic administration has only been shown to be effective in a mouse model of ischemia–reperfusion acute kidney injury |
| MitoQ [189,190] |
Mitochondrial oxidative stress |
Targets mitochondria to prevent lipid peroxidation |
Validation of phenotypic improvement in mouse aging model and chronic renal failure patients, large-scale clinical randomized controlled trials were lacking |
| Mito-TEMPO [191,192] |
Mitochondrial oxidative stress |
SOD enzyme mimics targeting ROS-mediated hypermethylation of the NDRG2 promoter |
Validation of phenotypic improvement in mouse UUO model and rat 5/6 nephrectomy CKD model |
| SS-31 [197,198,199,200] |
Mitochondrial oxidative stress |
Targets the inner mitochondrial membrane to scavenge mitochondrial oxygen radicals by tyrosine or dimethyltyrosine residues |
Validation of phenotypic improvement in rat 5/6 nephrectomy and UUO model |
| SS-20 [201] |
Mitochondrial oxidative stress |
Targets the inner mitochondrial membrane to scavenge mitochondrial oxygen radicals by tyrosine or dimethyltyrosine residues |
Validation of phenotypic improvement in mouse 5/6 nephrectomy model |
| mtCPP-1 [203] |
Mitochondrial oxidative stress |
Targets mitochondria to scavenge mitochondrial oxygen radicals by dimethyltyrosine residues |
Lack of animal model validation |