1. Factors Effecting the Photocatalytic Performance
In the context of organic synthesis, the manipulation of the reaction condition is pivotal for achieving the desired product with an optimal yield. To unlock the full potential of PNCs for their photocatalytic activity, it is imperative to comprehensively assess various aspects, including the impact of PNC size and bandgap tuning on its photocatalytic performance, interaction with solvents, and considerations related to acidity, ions, and moisture.
1.1. Effect of Size of NCs on Photocatalytic Activity
The use of semiconductors as photocatalysts requires them to exhibit a high absorption coefficient and ensure the photogenerated charge carriers reach the surface promptly with a minimum recombination. In addition, a suitable band alignment is crucial to enable the efficient charge transfer for oxidation and reduction processes. The use of small NCs is pivotal to achieving these objectives; their diminutive size facilities the swift transport of the photogenerated charge carriers to the surface. Semiconductor NCs exhibiting QCE are particularly promising candidates for photocatalysis.
1.1.1. Size Dependent Photocatalytic Activity of Lead Halide PNCs
It is widely recognized that strong QCE occurs when the NC size is smaller than the Bohr radius of the material. The Bulk Bohr exciton radius has been calculated using the equation 𝑎𝑏=𝜀𝑟(𝑚0/µ)𝑎0
, where 𝑎𝑏 is the exciton Bohr radius, 𝜀𝑟 is the relative dielectric constant of the bulk semiconductor, 𝑚0 is the mass of a free electron, µ is the reduced mass of the exciton, and 𝑎0
is the Bohr exciton radius of the hydrogen atom (0.529 Å). The calculated Bohr radius for CsPbBr
3 is 3.5 nm. The calculated Bohr exciton radius for CsPbBr
3 is 3.5 nm
[1][2].
Figure 1 shows the dependence of the UV–visible and photoluminescence (PL) spectra of different-sized CsPbBr
3 NCs intended for Csp
3-Csp
3 and Csp
3-Nsp
3 bond formation and their respective yields.
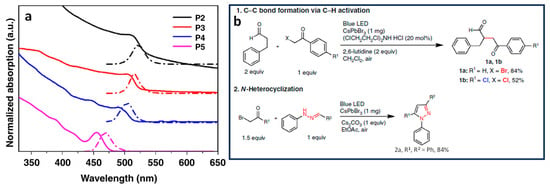
Figure 1. (a) UV–visible absorption and PL spectra (dashed lines) of different-sized CsPbBr3 NCs, P2-P5; (b) formation of Csp3-Csp3 and Csp3-Nsp3 bonds shown in red colour and their respective yields in reaction 1 and 2.
CsPbBr3 NCs, under quantum confinement, exhibit size-dependent properties. Zhu et al. conducted a study on the catalytic performance of organic compounds using different sizes of CsPbBr3 NCs. They prepared octylamine-capped NCs with different edge lengths, which exhibited bulk-like NCs from 2 to 100 nm (P1) and quantum-confined NCs, such as oleylamine-capped NCs with sizes of 14, 9, 6, and 4 nm (P2, P3, P4, and P5, respectively). Figure 1a shows the size-dependent absorption and PL of the different-sized CsPbBr3 NCs and their use as photocatalysts for Csp3-Csp3 bond formation via C–H bond activation (see reaction 1 and 2 in Figure 1b).
The results showed that smaller PNCs exhibited a higher initial activity for C–H bond activation, leading to the formation of product 1a in Figure 1b. Interestingly, P2–P4 NCs achieved product yields of 54–64% within 40 min, but the prolonged reaction time did not increase the yield significantly. On the other hand, P1 initially showed a slower production of 1a in Figure 1b but improved the yield up to 85% with a prolonged reaction time. This disparity was attributed to the small NCs, whose higher surface area-to-volume ratio enabled a swifter reaction rate during the initial stages. Moreover, it was observed that moisture in the solvent interacted more substantially with the smaller NCs as opposed to the larger ones, this resulting in progressive perovskite degradation and, hence, activity decrease. In addition, P5 exhibited a yield of 8%, primarily owing to its limited absorption of visible light and its increased susceptibility to degradation. In contrast, PNCs were stable when the reactions were performed under a pre-dried, non-halide solvent as ethyl acetate. Product 2a in Figure 1b was produced in an 87% and 86% yield with P1 and P2 after 6 and 2 h of reaction, respectively. This underscores the pivotal role of smaller-sized NCs in achieving high yields in shorter time frames.
1.1.2. Size Dependent Photocatalytic Activity of Lead-Free Halide PNCs
To the best of our knowledge, the systematic study of quantum-confined, size-dependent photochemical activity has not yet been explored using A
3M
2IIIX
9 and A
2M
IM
2IIIX
6 type NCs
[3]. Moreover, A
4M
IIM
2IIIX
12 type NCs, specifically Cs
4ZnSb
2Cl
12 LDP NCs, showed size-dependent optical properties. The calculated Bohr diameter of Cs
4ZnSb
2Cl
12 was 5.5 nm or above (due to
µchanges with different crystal facets). Caruso et al. reported different-sized organic capped Cs
4ZnSb
2Cl
12 NCs, namely 6.2 nm, 8.3 nm, 11.0 nm, and 15.6 nm NCs in the form of spherical dots
[4].
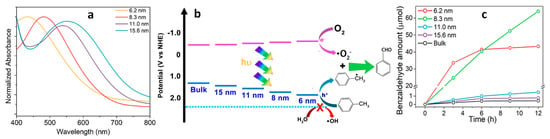
Figure 2a displays the size-dependent absorption features of Cs
4ZnSb
2Cl
12 NCs and, consequently, the bandgap tuning with the QCE. The authors conducted calculations to determine the CBM and VBM for bulk and various-sized Cs
4ZnSb
2Cl
12 NCs, as presented in
Figure 2b. Importantly, QCE was observed to fine-tune the edges of the conduction and valence bands. Notably, the shift in the VBM proved to be more significant than that of the CBM, thus providing an appropriate band alignment conducive to driving the oxidation of toluene. As shown in
Figure 2c, smaller-sized NCs exhibited a higher photocatalytic performance than bulk Cs
4ZnSb
2Cl
12. Specifically, the NCs with sizes of 6.2 nm and 8.3 nm exhibited superior performance when compared to larger-sized NCs. Notably, the 6.2 nm NCs displayed the highest activity during the initial 6 h, but their activity declined over time when compared to that of the 8.3 nm NCs. This reduction in activity can be attributed to the gradual degradation of the 6.2 nm NCs when exposed to water released during the prolonged benzaldehyde formation process.
Figure 2. (a) UV–vis absorption spectrum of various-sized Cs4ZnSb2Cl12 layered double perovskite (LDP) NCs; (b) scheme of the energy levels of the conduction and valence band edges for bulk and different-sized Cs4ZnSb2Cl12 NCs and mechanism of photocatalytic toluene oxidation to benzaldehyde formation; (c) time-dependent photo catalytic activity of benzaldehyde formation using bulk and NCs of Cs4ZnSb2Cl12. Reproduced with permission. Copyright: 2023, American Chemical Society.
In general, both Pb and Pb-free PNC-based photocatalytic reactions have shown that smaller-sized NCs with suitable light absorption characteristics exhibit high photocatalytic activity. However, this increased activity often increases at the cost of compromising their stability over extended periods. Therefore, it is vital to enhance the stability of smaller NCs for performing chemical reactions under ambient/water conditions. One potential approach involves the application of a thin SiO
2 layer to shield the perovskite NCs, as suggested in previous studies
[5][6]. Another strategy entails the incorporation of these smaller PNCs into the pores of stable materials, such as melamine foam, to address the stability concerns
[7]. Further investigation is warranted to comprehensively assess the photocatalytic performance and stability of these composite materials across a range of chemical reactions.
1.2. Stability and Reaction Condition Tolerance
In the context of organic synthesis using photocatalysis, the evaluation of perovskite materials becomes crucial, considering factors such as the co-catalyst, atmosphere, and type of solvent.
1.2.1. Role of a Co-Catalyst
CsPbBr3 NCs have been used in different reaction mediums to investigate the impact of ion effects and the role of solvents in perovskite photocatalysis. Reaction 1a in Figure 1b involves the use of (ClCH2CH2Cl)2NH2Cl as a co-catalyst with CsPbBr3 NCs photocatalyst, resulting in the formation of CsPbBrxCl3−x NCs. However, it is worth noting that the co-formation of excess Br ion during reaction 1a in Figure 1b could potentially lead to the subsequent Cl ion exchange with CsPbBrxCl3−x, thus resulting in the formation of the more stable CsPbBr3 perovskite. Conversely, reaction 1b in Figure 1b, which used Cl-substrates, demonstrated the complete transformation of CsPbBr3 NCs to CsPbCl3 NCs after the reaction. Furthermore, when non-halide solvents were employed in reaction 2a in Figure 1b, the overall stability of the PNCs (P1 and P4) was enhanced, thus improving the perovskite recyclability.
1.2.2. Role of Acidity
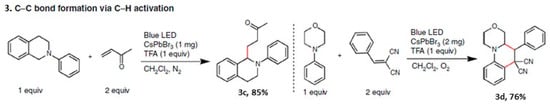
The role of acidity in perovskite photocatalysis is significant. When various organic acids, such as propionic acid, benzoic acid, or trifluoroacetic acid (TFA), are used as co-catalysts or substrates, an enhancement in PL was observed for P1 CsPbBr3 NCs up to an optimum concentration. This enhancement has been attributed to the carboxylic acid acting as a capping ligand through strong hydrogen bonding with the halide ions and to a robust interaction between the carboxylic acid and Pb atoms. Interestingly, the optimized concentration of TFA, which exhibits high PL intensity, also led to maximum product yields for specific reactions 3c and 3d in Figure 3. Therefore, non-halide organic acids not only stabilize the PNCs but also enhance the overall catalytic efficiency.
Figure 3. C–C bond product formation (shown in red colour bonds in the products 3c and 3d) and respective chemical yields.
1.2.3. Role of Air
Air tolerance is an important aspect in chemical synthesis. While molecular photocatalysts typically require air-free conditions, PNCs do not. In fact, PNCs exhibit strong quenching with organic substrates when compared to oxygen (O2). In contrast, molecular photocatalysts often show significant and competitive O2 quenching, thus resulting in a poor catalytic performance. Consequently, PNCs demonstrate catalytic performance one to two orders of magnitude higher for reactions 1, 2, and 3 (in Figure 1b and Figure 3) than that of CdSe and molecular photocatalysts, making them highly efficient in air-tolerant reactions.
1.3. Role of Bandgap Tuning of Perovskite NCs on Photocatalytic Organic Reactions
Bandgap tuning plays a crucial role in selectively activating organic substrates during photocatalytic reactions. Three strategies can be used to tune the bandgap of metal halide PNCs (AM
IIX
3), specifically engineering the A- and M
II-site cations and X-site anion. Out of these three strategies, halide exchange has been promising for tuning the bandgap
[8].
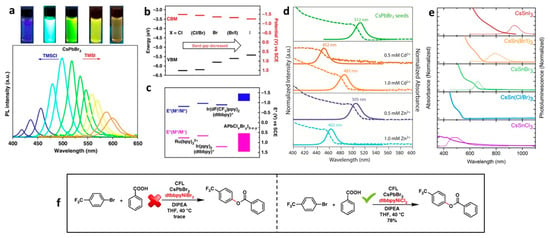
Figure 4a shows the colour and PL tuning across the entire visible range of lead halide PNCs by treating the CsPbBr
3 NCs with chloride and iodide precursors.
Figure 4b shows the band alignment of CsPbX
3 NCs (X: Cl, Br, and I). When the X changes from Cl to I from Br, different energy levels can be accessed, such as the well-known molecular catalysts. This allows for the activation of some interesting photochemical reactions, specifically for C–O bond formation. When CsPbBr
3 NCs were used in combination with dtbbpyNiBr
2 as a co-catalyst, only a minimal amount of product was generated, as shown in the reaction in
Figure 4f. In contrast, when dtbbpyNiCl
2 was present alongside CsPbBr
3, a 78% yield was achieved. This indicates that the anion exchange process, wherein Br is replaced by Cl, shifts the PNC CBM to a higher energy, as shown in
Figure 4b. This shift eases energetically favourable electron transfer processes during the reaction. Substantiating this mechanism, the study observed efficient triplet energy transfer from Ir(bpy)
3 molecular photocatalysts to the substrates, a result achieved through the modification of substituent groups.
Figure 4. (
a) Tunability of photoluminescence (PL) spectra of CsPbBr
3 NCs using anion exchange with trimethylsilyl chloride or iodide and their photographs under UV illumination; (
b) band edges of APbCl
xBr
yI
3−x−y; (
c) excited state potential (E*) range of APbCl
xBr
yI
3−x−y comparing with noble-transition metal photocatalysts; (
d) absorption (dashed lines) and PL spectra of Zn- and Cd-doped CsPbBr
3 NCs. Reproduced with permission
[9]. Copyright: 2017, American Chemical Society; (
e) absorption and PL spectra (dashed lines) of CsSnX
3 NCs. Reproduced with permission
[10]. Copyright: 2016, American Chemical Society; (
f) C–O bond formation with perovskite band-tuning.
B-site engineering of lead halide PNCs (LHPNCs) leads to the tuning of the bandgap in a relatively lower range than halide exchange. For instance, in CsPbBr
3 NCs, a partial substitution of Pb
2+ with a smaller transition metal ions, such as Zn
2+ and Cd
2+, would increase the bandgap by ~40–60 nm, as shown in the
Figure 4d, by contracting the lattice
[9]. On the other hand, the introduction of lanthanide metal ions with a +3 oxidation state (e.g., Ce
3+, Eu
3+, Sm
3+, Tb
3+, Dy
3+, Er
3+, and Yb
3+)
[11][12][13] and a transitional Mn
2+ ion
[13][14] leads to multicolour emission peaks related to dopant and hosts emission peaks spanning from visible to the near infra-red (NIR) region by creating new luminescent centres but with slightly less or no changes in the bandgap of the host NCs
[12]. In contrast, the complete replacement of Pb
2+ with Sn
2+ (CsSnX
3) allow for the bandgap tuning of lead-free PNCs in the visible and NIR region, as shown in
Figure 4e. However, the photocatalytic performance of these doped and CsSnX
3 NCs for organic transformations are yet to be explored
[15]. Furthermore, the halide exchange strategy can also be extended to other lead-free perovskites NCs A
3M
2IIIX
9 and A
2M
IM
IIIX
6 to tune the bandgap
[16][17].
The influence of the A-site cation on the bandgap in MHP is generally minimal, as the A-site cation does not directly participate in shaping the electronic band structure. However, slight adjustments in the bandgap can be achieved by considering the size differences of the A-site cation, which may lead to alterations in the octahedral tilting and/or lattice contraction or expansion
[18][19]. Hazarika et al. demonstrated that introducing Cs into FAPbI
3 NCs resulted in the fine-tuning of absorption and emission properties, shifting them from 800 nm to 650 nm
[20].
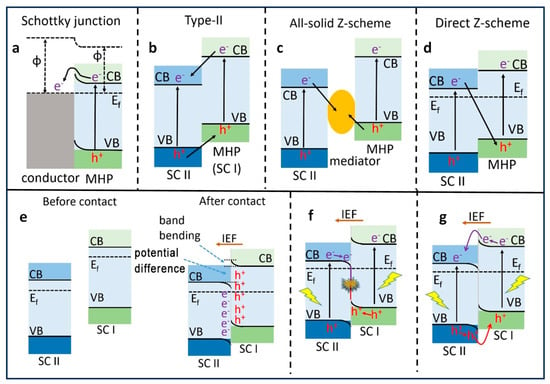
2. Types of Heterojunctions for Photocatalysis
The superior photocatalytic performance of PNCs compared to bulk perovskite is evident. Nevertheless, the heightened Coulombic attraction force between opposite charge carriers within smaller NCs often results in a pronounced recombination, thereby hampering the migration of charge carriers. To ameliorate this issue and enhance the separation of electron–hole pairs, leading to an improved photocatalytic performance and increased stability of PNCs, diverse heterostructures have been innovatively engineered. These heterostructures incorporate varying band alignments, encompassing Schottky, type-II, and Z-scheme heterojunctions, thereby opening new avenues for advanced materials and enhanced photocatalysis. These heterostructures have been prepared by combining semiconductor–conductor and semiconductor (SC I)–semiconductor (SC II) with unequal band energy levels.
Various metals, such as Au, Ag, Pt, and carbon-based materials, such as graphene and reduced graphene, possess Fermi energy levels that are relatively positive in comparison to perovskite materials. When these conductive materials are in contact with the perovskite materials, electrons are transferred from the semiconductor to the metal until equilibrium is reached. This electron transfer leads to an upward shift in the energy bands of the semiconductor at the interface, thus resulting in band bending, as illustrated in
Figure 5a. This type of junction is commonly referred to as a Schottky junction, and the band bending it creates gives rise to an energy barrier known as the Schottky barrier. The magnitude of the band bending and the Schottky barrier depends on the difference in work function between the metal and semiconductor
[21]. It is important to note that the effect of band bending is less pronounced when dealing with semiconductor NCs in contrast to bulk semiconductors. Consequently, establishing Schottky junctions with NCs is considered a promising approach to enhance photocatalysis. Schottky heterojunctions serve a dual purpose in this context; they not only facilitate the extraction of charge carriers by promoting charge separation, but they also act as a co-catalyst, offering active catalytic sites essential for specific photocatalytic processes
[22].
Figure 5. Schematic illustration of the band alignments of several possible heterojunctions. (
a) Schottky heterojunction; (
b) type-II heterojunction; (
c) all-solid-z-scheme hetero junction; (
d). direct Z-scheme heterojunction; (
e) the relative band positions and Fermi level of semiconductor (SC) I and SC II before and after contact; (
f,
g) After illumination with light, the charge migration follows the Z-scheme and type-II pathways. VB: valence band; CB: conduction band; E
f: Fermi level (dashed line); IEF: internal electric field. Reproduced with permission
[23]. Copyright: John Wiley and Sons.
When two semiconductors SC I and SC II, each having differing CB and VB energies, come into contact, they can form a type-II heterojunction, as depicted in Figure 5b. This unique type-II band alignment enables the transfer of photogenerated electrons from SC I to SC II and photogenerated holes from SC II to SC I. Recent studies have extensively explored type-II band alignment through the creation of heterojunctions involving perovskites and various semiconductors, such as TiO2, graphitic carbon nitride (g-C3N4), CdS, etc. Depending on the specific characteristics of the semiconductors involved, some of these heterojunctions may even result in the formation of p-n heterojunctions. When p-type and n-type semiconductors with differing Fermi energy levels combine to form a junction, the flat bands of n-type and p-type undergo upward and downward band bending, respectively, at the interface. This thermodynamically favourable band alignment eases the separation of photogenerated electrons and holes, causing them to reside on different sides in distinct semiconductors, thus enhancing the electron and hole separation and thereby increasing photocatalytic performance.
To expedite the separation and transfer of photogenerated electrons and holes, a commonly employed approach is the utilization of a type-II band alignment. However, in type-II heterojunctions, the redox potential of the photocatalyst is often compromised due to oxidation and reduction processes occurring on the semiconductor with lower oxidation and reduction potentials, respectively
[24]. In contrast, a Z-scheme heterojunction can maximize the redox potential of the heterostructures while maintaining efficient light absorption
[25]. This proves particularly advantageous in cases where high reduction potential CB and oxidation potential VB are required, such as in a water splitting reaction. The band alignment in Z-scheme heterojunctions closely resembles that of type-II heterojunctions, with a key distinction being the direction of photogenerated electron transfer from SC II to SC I as illustrated in
Figure 5c, d. Initially, all-solid Z-scheme heterojunctions where two different semiconductors were connected via an electron mediator, such as Ag, Au, and Pt, were reported. Under illumination, photoinduced electrons are transferred from the CB of SC II to VB of SC I through the electron mediator (
Figure 5c). Consequently, the photogenerated holes and electrons in SC II and SC I are spatially separated and can be harnessed for various photocatalytic applications. However, the use of such expensive materials as electron mediators restricts their large-scale application in photocatalysis. Subsequently, direct Z-scheme heterojunctions were developed, where two different semiconductors with varying Fermi levels were in direct contact with each other. This results in charge distribution at the interface under dark conditions, causing band bending and the creation of an interfacial built-in electric field (IEF) in direct Z-scheme heterojunctions (
Figure 5d). Moreover, a substantial difference in work function between SC I and SC II results in the formation of relatively large barriers that hinder electron transfer from SC I to SC II and hole transfer from SC II to SC I, defining the formation of a direct Z-scheme heterojunction. However, when the difference of work functions of SC I and SC II is smaller, a type-II band alignment can still co-exist with a Z-scheme heterojunction. Researchers have demonstrated the formation of both type-II and Z-scheme heterojunctions using the same perovskite materials. For instance, Tüysüz et al.
[26] prepared a type-II heterojunction with CsPbBr
3/TiO
2, while Yu et al. established a Z-scheme heterojunction
[27]. It appears that two different semiconductors with n-type characteristics can form a Z-scheme heterojunction.
3. Carbon–Carbon (C–C) Bond Formation
C–C bond formation is a foundational process in organic synthesis, crucial for creating complex molecular structures
[28]. Traditional methods, including artificial photoredox catalysis, have advanced, but they often require noble metals or complex procedures
[29][30]. Addressing this, NCs emerge as promising photocatalysts for C–C bond formation under visible light. Unlike conventional systems, PNCs offer simplified product separation, eliminating the need for costly noble metal catalysts. Their unique advantage lies in an efficient energy transfer to organic substrates, initiating photocatalytic reactions with long-lived molecular triplets. This introduces a tuneable and versatile photocatalyst, showcasing PNCs as a potential game-changer for sustainable and efficient C–C bond-forming reactions under mild conditions.
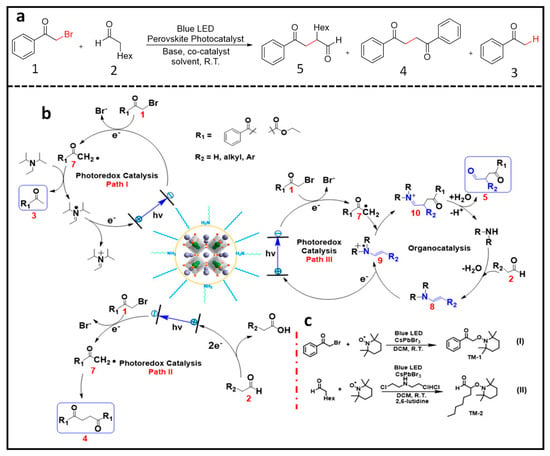
3.1. α-Alkylation of Aldehydes Using LHPNCs
Zhu et al. demonstrated the notable selectivity and high yield achieved in the α-alkylation of aldehydes. This process facilitated the formation of C–C bonds and reduction of C–Br using CsPbBr
3 and MAPbBr
3 nanoparticles (NPs) as photocatalysts under blue LED irradiation
[31]. Impressively, they reported an exceptionally high turnover number (TON) exceeding 52,000, surpassing the efficiency of catalysts based on noble metals. In this method, octylamine-capped CsPbBr
3 NPs (P1) were used. Under blue LED illumination in an open-air environment and in the presence of CsPbBr
3 NPs, dicyclohexylamine as a co-catalyst, and 2,6-lutidine as a base, compounds
1 and
2 (
Figure 6a) yielded compounds
3,
4, and
5 (
Figure 6a). The formation of these three compounds was explained by a radical mediated mechanism involving three pathways. Upon perovskite photoexcitation, the electron is transferred from compound
1 and is reduced, and it eventually forms radical 7 (
Figure 6b). In the presence of a sacrificial donor, N,N-diisopropylethylamine (DIPEA), radical 7 transforms into compound
3 (path I in
Figure 6b). In the absence of a donor, radical 7 self-couples, resulting in the formation of compound
4 (path II in
Figure 6b). The regeneration of the photocatalyst occurs through the oxidation of the electron donor and aldehyde in path I and path II, respectively. In path III (
Figure 6b), oxidative quenching with in-situ generated enamine 8 (formed by the reaction between dicyclohexylamine and octanal with a loss of a water molecule) leads to radical cation 9, which, upon reaction with radical 7 followed by hydrolysis, generates the iminium cation 10, which releases product 5 (Path III in
Figure 6b) and regenerates the co-catalyst. Additionally, radical trapping experiments were performed to confirm the formation of radical intermediates using TEMPO, as shown in
Figure 6c.
Figure 6. (
a) Photocatalytic reductive dehalogenation, sp
3-C coupling, and α-alkylation of aldehydes; (
b) proposed mechanism for perovskite catalyzed dehalogenation, sp
3 carbon coupling, and α-alkylation; (
c) TEMPO trapped experiment for radical intermediate validation. Reproduced with permission
[31]. Copyright: 2019, American Chemical Society.
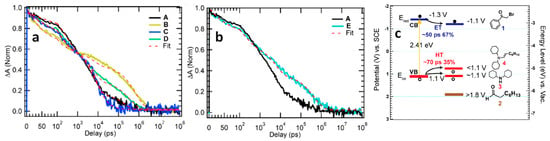
To gain a deeper understanding of the reaction mechanism, Wang et al. employed transient absorption (TA) spectroscopy to investigate the charge transfer and reaction dynamics, focusing specifically on the selective formation of product 5 (
Figure 6a)
[32]. In this study, amine-free CsPbBr
3 NCs were utilized as photocatalysts to avoid a potential charge transfer to the amine ligand.
Figure 7a shows the results of the ground-state bleach dynamics for different systems: pure CsPbBr
3NCs (A), CsPbBr
3 NCs + 2-bromoacetophenone (B), CsPbBr
3 NCs + octanal (C), CsPbBr
3 NCs + octanal + dicyclohexylamine (D), and CsPbBr
3 NCs + dicyclohexylamine (E). The analysis revealed that both B, D, and E (
Figure 7a,b) exhibited decay times of ~50 ps, ~70 ps, and ~70 ps, respectively, followed by a slower decay pattern compared to pure CsPbBr
3 NCs (A).
Figure 7c provided insights into the reduction and oxidation potentials of the reactants, photocatalysts, and co-catalysts. The energy level of 2-bromoacetophenone was favourable for ultra-fast electron transfer, occurring within ~50 ps. On the other hand, dicyclohexylamine and the in situ-formed enamine facilitated the hole transfer, with a timescale of ~70 ps. However, it was determined that a hole transfer to dicyclohexylamine did not result in the desired oxidized enamine intermediate, eliminating this possibility from the mechanism. Consequently, following the ultrafast electron and hole transfer processes, charge-separated states were formed with a lifetime of approximately 0.8 µs. This 0.8 µs charge-separated state allows the photogenerated-charged radical intermediates to undergo C–C bond formation through a biradical pathway
[32].
Figure 7. (a) Normalized transient absorption (TA) kinetics probed at the centre of the NCs exciton bleach spectrum for A–D, red-dashed traces are fits to kinetics; (b) normalized TA kinetics and fits for A and E where A, B, C, D, E represent pure CsPbBr3 NCs, CsPbBr3 NCs + 2-bromoacetophenone, CsPbBr3 NCs + octanal, CsPbBr3 NCs + octanal + dicyclohexylamine, CsPbBr3 NCs + dicyclohexylamine, respectively; (c) electrochemical potential and energy level for reactants and CsPbBr3 NCs. Reproduced with permission. Copyright: 2020, American Chemical Society.
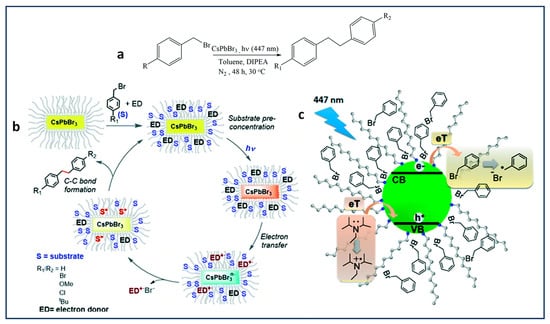
3.2. Benzyl Bromide Coupling Using LHP NCs
Pérez-Prieto and co-workers have demonstrated that the use of dodecylamine-capped CsPbBr
3 NCs, combined with an electron donor, eventually results in photoredox-catalysed homo- and cross-coupling of benzyl bromides under visible light (447 nm) excitation, as shown in
Figure 8a
[33]. In this study, the authors found the thermodynamically unfavourable band alignment for electron transfer between benzyl bromide (−1.5 V vs. SHE) and CBM of CsPbBr
3 NCs (−0.96 V vs. SHE). This, in principle, unexpected result was rationalized as due to substrate pre-concentration at the NC surface, facilitated by the van der Walls interaction between the benzyl bromide and alkyl chains of the organic ligand anchored to the NC surface, followed by an electron transfer from the NC to the benzyl bromide, thus eventually generating a high concentration of benzyl radicals close to or at the NC surface and driving the C–C coupling reaction forward.
Figure 8. (a) Photocatalytic aryl bromide coupling reaction; (b) schematic representation of the cooperative action between the NC surface and the capping for the catalysed coupling reaction; (c) the process occurring after excitation of the CsPbBr3 NCs by vis-lamp in the presence of benzyl halide as electron acceptor and DIPEA as electron donor within the capping.
Moreover, the hole in the valence band of CsPbBr3 refilled by an electron transfer from DIPEA facilitated by the favourable HOMO level (−5.76 V) relative to the VBM of CsPbBr3 (between −5.85 to −6.4 V), as shown in the Figure 8c. Notably, a TON of 17,500 with a >80% product yield was achieved for the C–C coupling of benzyl bromide. Additionally, the authors discovered that the product yield is influenced by substitution at the para position, with the yield being H, tBu, OMe > Cl > Br.
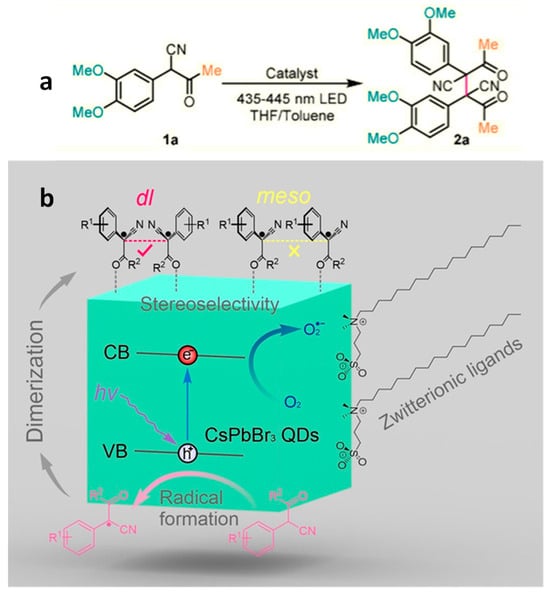
3.3. Stereo Selective C–C Oxidative Coupling Reaction
Chen and co-workers demonstrated the first photocatalytic stereoselective C–C oxidative coupling reaction using PNCs
[34]. They used 2-(3,4-dimethoxyphenyl)-3-oxobutanenitrile (compound
1a in
Figure 9a) as a model substrate and CsPbX
3 (X = Br, I) NCs as photocatalyst under visible light illumination. This study revealed the critical role of surface ligands ensuring a stable reaction. In this context, CsPbX
3 NCs capped with oleic acid/oleylamine (OA/OAm) exhibited impressive results, with a yield of 95% and excellent stereoselectivity of 99%. However, a significant drawback was the rapid degradation of OA/OAm-capped CsPbX
3 NCs during the photocatalytic reaction. This issue was attributed to the instability of CsPbI
3 NCs and possible ligand detachment during the post-reaction purification process. To address this challenge, the authors introduced surface modification to transform OA/OAm-CsPbBr
3 NCs into zwitterionic (ZW) ligand-capped CsPbBr
3 NCs. This innovation not only improved the recyclability of the reaction, extending its usability to at least three cycles, but also enhanced the reaction kinetics. This enhanced reactivity was explained by the reduction in the density of surface ligand coverage, decreasing from 5.4 nm
2 for OA/OAm-capped CsPbBr
3 NCs to 3.0/nm
2 for ZW- capped CsPbBr
3 NCs. This reduction facilitated increased substrate adsorption onto the NC surface and lowered the kinetic barrier for the reaction. It is worth noting that ZW ligands strongly adhered to the PNC surface, which enabled multiple washing steps during the NC purification process to remove excess ligands. This feature is advantageous for optoelectronic applications as well
[35][36].
Figure 9. (a) Photocatalytic stereo-selective C–C oxidative coupling reaction of 2-(3,4-dimethoxyphenyl)-3-oxobutanenitrile; (b) proposed reaction scheme for the stereoselective dimerization of α-keto nitriles photocatalyzed by zwitterion capped CsPbBr3 NCs. Reproduced with permission. Copyright: 2020, John Wiley and Sons.
Furthermore, the scope of the reaction was expanded by introducing various substituents at different positions on the phenyl ring. They discovered that electron donating groups in the para position plus an extended conjugated π system were essential for the C–C coupling (dimerization) reaction to occur. Figure 9b illustrates the reaction mechanism, which begins with the formation of the substrate (α-aryl keto nitriles) radical due to the oxidation of photoexcited holes in the PNCs and follows with the C–C bond coupling of neighbouring radicals. Depending on the relative configuration of the two radicals on the NC surface, either dl- or meso-isomers can be formed. However, less steric hindrance between the aryl groups of the radicals in the trans-arrangement and an attractive interaction between the gauche cyano groups upon molecular structural relaxation drives the reaction kinetically and thermodynamically to the formation of the dl-isomer over the meso-isomer.
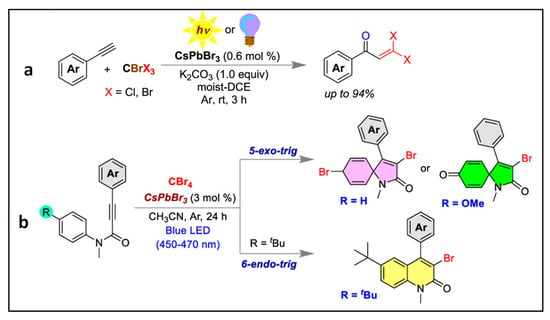
3.4. Synthesis of Gem-Dihaloenones by the Activation of C-Br Bonds of CBrX3 (X = Cl, Br)
Mal et al. employed CsPbBr
3 NCs (PLQY of 93%) for the synthesis of gem-dihaloenones via the activation of C–Br bonds of CBrX
3 (X = Cl, Br) as shown in
Figure 10a, thus achieving a 94% yield of the enone under sunlight
[37]. The same group reported chemo-divergent reactions (i.e., obtention of different products by changing the reaction conditions), such as the functionalization of N-methylalkanamides to form 6-endo-trig or 5-exo-trig mode of cyclization via C–Br bond activation of CBr
4 using CsPbBr
3 NCs obtained from an unprecedented bromide precursor dibromoisocyanuric acid as shown in
Figure 10b. In both cases, an electron transfer takes place from the excited photocatalyst to the reactant to generate a radical anion intermediate.
Figure 10. (a) Aliphatic C–Br bond activation of CBrX3 (X = Br, Cl) using the CsPbBr3 NCs. Reproduced with permission. Copyright: 2023, American Chemical Society; (b) chemodivergent synthesis of 3,8-dibromo-1-methyl-4-phenyl-1-azaspiro[4.5]deca-3,6,9-trien-2-on or 3-bromo-1-methyl-4-phenyl-1-azaspiro[4.5] deca-3,6,9-trien-2,8-dione or 3-bromo-6-(tert-butyl)-1-methyl-4-phenylquinolin-2(1H)-one using CsPbBr3 NCs photocatalyst under visible light irradiation. Reproduced with permission. Copyright: 2023, American Chemical Society.
3.5. Aminomethylation of Imidazo-Fused Heterocycles
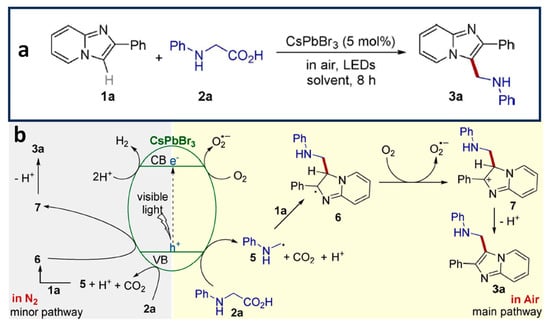
Shi et al. have reported the direct aminomethylation of imidazo-fused heterocycles through a decarboxylative coupling reaction of N-phenyl glycine (2a in
Figure 11a) and imidazo-fused heterocycles (1a in
Figure 11a), using octylamine-capped CsPbBr
3 NCs (P1)
[38]. This reaction was carried out under open-air conditions at room temperature and with a high yield (>90%). The mechanism behind this reaction involves radical intermediates. Initially, an electron transfer from 2a (in
Figure 11a) to the generated hole results in the formation of intermediate radical 5 (in
Figure 11b), accompanied by the release of carbon dioxide and H
+. Subsequently, the addition of radical 5 (in
Figure 11b) to the C=C bond of 1a (in
Figure 11a) leads to the formation of radical 6 (in
Figure 11b), followed by oxidation to generate radical 7 (in
Figure 11b). Eventually, radical 7 (in
Figure 11b) undergoes deprotonation to yield the desired product 3a (in
Figure 11b). Importantly, the CsPbBr
3 NCs exhibited excellent recyclability, enabling the reproduction of the product with a high yield for up to five cycles.
Figure 11. (a) Photocatalytic aminomethylation of imidazo-fused heterocycles; (b) plausible mechanism of aminomethylation of imidazo-fused heterocycles using CsPbBr3 NCs under the irradiation of visible light. Reproduced with permission. Copyright: 2020, John Wiley and Sons.
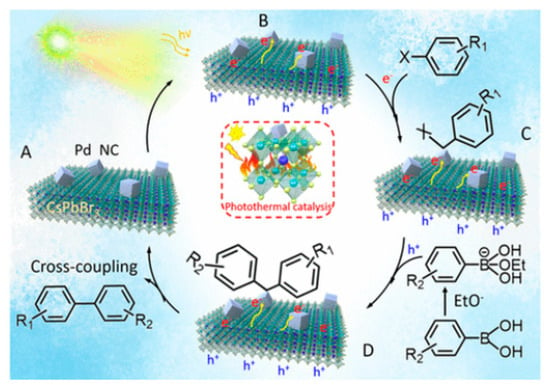
3.6. Photothermal Suzuki Coupling CsPbBr3/Pd
In organic synthesis, the Suzuki coupling reaction is a pivotal method for C–C bond formation. Initially, thermally driven Suzuki coupling reactions utilized Pd complexes as homogeneous catalysts to perform these reactions. Over time, diverse heterogeneous catalysts have been developed to improve such coupling reaction. This was achieved through the integration of photocatalysts, combining Pd NPs with materials such as TiO
2, SiC, C
3N
4, and WS
2 [39][40][41][42][43]. In this context, Roeffaers et al. employed Pd nanocubes deposited onto CsPbBr
3 NCs as an efficient photothermal catalyst for Suzuki coupling reaction of iodobenzene with phenylboronic acid as a model reaction under solar irradiation (AM 1.5 G) at 30 °C for 4 h
[44]. The 3 wt% Pd/CsPbBr
3 photocatalyst exhibited the highest conversion, being 11-fold higher than when using CsPbBr
3 NCs; moreover, it occurred at a nearly 100% selectivity. Furthermore, the 3 wt% Pd/CsPbBr
3 catalyst displayed an excellent performance for several reactions involving different aryl halides and electron-withdrawing and electron-donating substituents.
Notably, this catalytic performance has been attributed to the synergistic effects of both photoinduced and thermal contributions, with the photothermal activity of 3 wt% Pd/CsPbBr3 outperforming both the photoinduced activity of pristine CsPbBr3 and the photo-thermal activity of Pd nanocubes at 60 °C, underscoring the unique advantages of this composite. Furthermore, this study elucidated the activation energies of 40.4 and 30.1 kJ mol−1 for the thermal and photothermal catalysts, respectively. This information helps to explain the favourable reaction conditions observed in the Suzuki coupling reaction under photothermal catalysis. Stability tests confirmed the sustained activity of the 3 wt% Pd/CsPbBr3 composite for up to 24 h.
Mechanistic studies corroborated the key role of alkoxides, such as EtO
-, in triggering the Suzuki coupling reaction. The plausible mechanism involves photoinduced electrons in CsPbBr
3 transferring to Pd through the Schottky contact
[45] as shown in the
Figure 12A,B, thus resulting in an electron-rich Pd species that can activate the C–X bond of aryl halides, to produce Pd-adsorbed aryl intermediates (
Figure 12C). Simultaneously, phenylboronic acids and alkoxides lead to a boron alkoxide complex, which interacts with photoinduced holes, weakening the carbon–boron bond as shown in the
Figure 12C,D. As the activated phenylboronic acid complex migrates to the redox-activated aryl halide, the coupling reaction produces the corresponding biphenyl products (
Figure 12A–D).
Figure 12. Schematic representation of the sp2carbon coupling reaction of iodobenzene with phenylboronic acid using Pd/CsPbBr3 Schottky junction photocatalyst. Reproduced with permission. Copyright: 2022, American Chemical Society.
3.7. C–C Bond Cleavage via Decarboxylation and Dehydrogenation Reactions
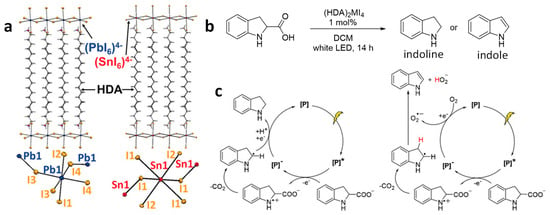
Soo and co-workers conducted a study involving the C–C cleavage through decarboxylation and dehydrogenation reactions using stable, structurally defined 2D lead and tin iodide perovskites
[46]. To enhance the stability of these iodide-based perovskites, they introduced hexadecyl ammonium (HDA) cations as the intercalating layers between the metal halide nanosheets, forming (HDA)
2MI
4 (M = Pb, Sn), as illustrated in
Figure 13a. It is important to note that the inclusion of the hydrophobic, long HDA cation greatly improves the stability of these (HDA)
2MI
4 perovskites in a polar solvent, such as water. However, the stability of (HDA)
2SnI
4 is somewhat lower than that of (HDA)
2PbI
4. This disparity primarily arises from the susceptibility of Sn
2+ to oxidize to Sn
4+. Both 2D perovskites, (HDA)
2PbI
4 and (HDA)
2SnI
4, exhibit distinct bandgaps, specifically at 2.3 and 1.9 eV, respectively. These bandgap values make them suitable candidates for visible light-driven photochemical reactions.
Figure 13. (a) Crystal structures of (HDA)2PbI4 and (HDA)2SnI4; (b) representative decarboxylation and dehydrogenation reactions catalyzed by the (HDA)2PbI4 and (HDA)2SnI4 perovskites; (c) suggested mechanism for the decarboxylation (left) and dehydrogenation reactions (right). [P] represents the perovskite. Reproduced with permission. Copyright: 2019, John Wiley and Sons.
The authors used these perovskites for decarboxylation and dehydrogenation of indoline-2-carboxylic acid to indoline and indole, respectively, in the presence of O2 (Figure 13b). Notably, (HDA)2PbI4 yielded higher quantities of indoline and indole compared to the lead-free (HDA)2SnI4. This difference in performance was attributed to the oxidative instability of Sn2+ and abundance of traps in the (HDA)2SnI4 material. Figure 13c provides an explanation of the proposed mechanism, suggesting that photooxidation is the rate determining step in both reactions, with the formation of a superoxide radical playing a crucial role in the dehydrogenation process.
4. Doped NCs
Doping serves as a highly effective and versatile strategy in photocatalysis, particularly notable in advancements with halide perovskite photocatalysts. The deliberate introduction of suitable transition metal dopants, through both bulk and surface doping, plays a pivotal role in optimizing photocatalytic processes, notably for CO
2 reduction and organic synthetic reactions
[47]. The success of this strategy lies in its ability to fine-tune material properties, enhancing stability, activity, and selectivity
[47]. In the case of CsPbBr
3, theoretical studies and experimental validations demonstrate that Co and Fe dopants with compatible lattice parameters significantly reduce energy barriers for key reaction steps, showcasing catalytic superiority
[48][49]. This enhanced activity is attributed to the adsorption of CO
2 at doping sites, optimizing the distance to the metal dopant. Experimental confirmation, such as with Fe-doped CsPbBr
3, further underscores the efficacy of doping in activating photochemical reactions, promoting superior product yields compared to undoped counterparts
[50].
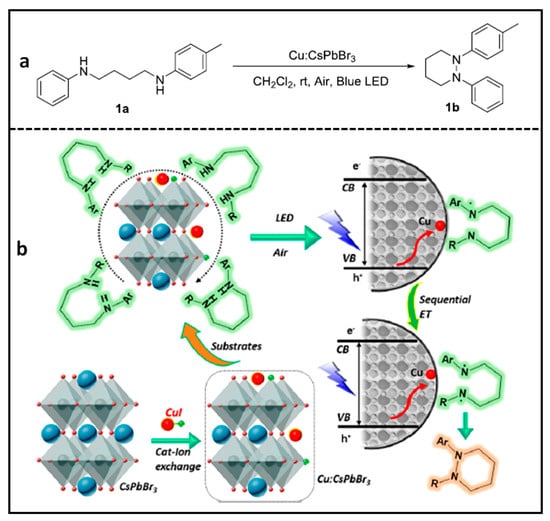
Cu-Doped CsPbBr3 NCs for N–N Heterocyclization Reaction
Yan et al. developed a novel photocatalyst that employs transition metal Cu(I) ~ 1% doped CsPbBr
3 NCs for a
N–N heterocyclization reaction
[51], which is a challenging reaction due to saturated sp
3 C-based
N–N rings, which are very labile in nature. The doping of Cu(I) was performed by replacing the Cs atoms on the surface of CsPbBr
3 NCs. Thus, surface bound Cu(I) ions allow for the accumulation of holes after photoexcitation owing to the fact that their energy levels lie above the VB of CsPbBr
3 of the NCs.
These Cu(I)-doped CsPbBr3 NC photocatalysts have been used to induce pyridazine formation from reactant 1a (in Figure 14a) with a 90% yield within 18 h, using the conditions mentioned in the reaction. In contrast with a single-carrier transfer in many catalytic reactions, in this reaction, photoinduced holes that were transferred to a diamine substrate form a biradical upon a consecutive inner sphere charge transfer from VB of the NC to surface bound Cu(I) orbitals. Therefore, a multi-charge transfer from the diamine substrate led to the formation of a radical intermediate, which is the key step for the intermolecular biradical formation. As a result, an oxidative intermolecular N–N coupling reaction takes place, as shown in the scheme in Figure 14b. It is worth mentioning that product 1b (in Figure 14a) cannot be formed when using a molecular photocatalyst or free Cu in a solution as a co-catalyst. Furthermore, this reaction can be applied to many other aliphatic and aromatic cyclic rings. This study highlights the potential applications of transition metal-doped PNCs in photocatalytic organic synthesis.
Figure 14. (a) Photocatalytic N–N heterocyclization reaction; (b) photocatalytic sequential electron transfer approach for di-radical path N-heterocyclization using Cu-doped CsPbBr3 NCs. Red circle represents the Cu(I)-doping in the perovskite crystal structure. Reproduced with permission. Copyright: 2021, American Chemical Society.
5. Photocatalytic Polymerization
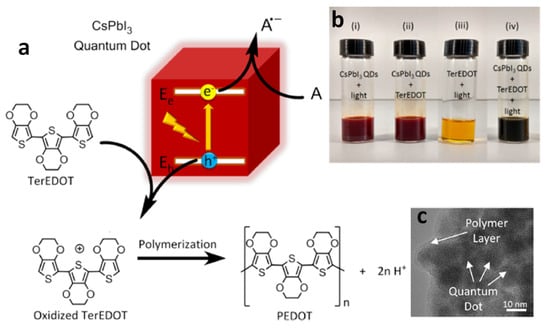
Poly(3,4-ethylenedioxythiophene) (PEDOT), a widely used conducting polymer in optoelectronic devices, is typically synthesized through the chemical or electrochemical oxidation of 3,4-ethylenedioxythiophene. However, integrating PNCs into a PEDOT matrix using conventional methods poses challenges, risking contamination or damage. Photodeposition, leveraging halide perovskites attributes, offers a clean approach. This process, facilitated by photoinduced electron transfer, enhances optoelectronic properties by efficiently transferring carriers between perovskite QD and the formed polymer matrix.
In this context, Tüysüz et al. were the first to report the photocatalytic polymerization of 2,2’,5’,2”-ter-3,4-ethylenedioxythiophene (TerEDOT) using CsPbI
3 NCs and visible light illumination
[52]. The favourable band alignment between TerEDOT and CsPbI
3 NCs enabled the transfer of photoexcited holes to TerEDOT, leading to the oxidation of TerEDOT and, ultimately, the formation of poly-(3,4-ethylenedioxythiophene) (PEDOT) in the presence of either benzoquinone or O
2 as an electron acceptor (
Figure 15a). It is noteworthy that, when this reaction is performed in the presence of O
2, the cubic structure of CsPbI
3 NCs transformed into an orthorhombic structure, which is unsuitable for optoelectronic applications.
Figure 15. (a,b) Illustration of the proposed mechanism for photocatalytic polymerization of TerEDOT over CsPbI3 NCs under visible light irradiation; (c) TEM image of CsPbI3 NCs encapsulated by PEDOT and CsPbI3 NCs. Reproduced with permission. Copyright: 2017, American Chemical Society.
Conversely, in the presence of 1,4-benzoquinone (BQ), the cubic structure of CsPbI
3 NCs is maintained. Furthermore, the resulting polymer acted as a protective layer, encapsulating the CsPbI
3 NCs and preserving their cubic structure (
Figure 15b). This study emphasizes the utilization of narrow bandgap perovskites for photocatalytic applications. Additionally, Chen and co-workers investigated a similar reaction using CsPbBr
xI
3−x NCs and observed that CsPbI
3 NCs outperformed CsPbBr
xI
3−x NCs in the photocatalytic polymerization of TerEDOT
[53]. This superior performance was attributed to the lower binding energy of CsPbI
3 NCs (152 meV) compared to CsPbBr
1.
5I
1.
5 NCs (472 meV) and CsPbBr
3 NCs (520 meV). Furthermore, they demonstrated that post-treatment with methyl acetate of oleic acid/oleylamine-capped CsPbBr
xI
3−x NCs accelerates the photocatalytic polymerization of TerEDOT.
6. Coupling of Thiols to Disulfides
Disulfides play a crucial role in facilitating the folding of proteins into biologically active conformations
[54]. Additionally, they find extensive applications in the fine chemical industry, serving as antioxidants, pesticides, pharmaceuticals, and vulcanization agents
[55]. The conventional method for obtaining symmetric disulfides involves oxidative coupling of thiols with oxidants
[56]. However, this process often presents challenges, including issues such as over-oxidation; the need for high catalyst-loading, excessive, and costly oxidants; reliance on strong acidic or basic media; and elevated temperatures. The quest for a mild, environmentally friendly, selective, and efficient thiol-coupling protocol remains a significant goal. In this regard, photocatalysis emerges as a gentler pathway to accomplish this crucial transformation
[57][58].
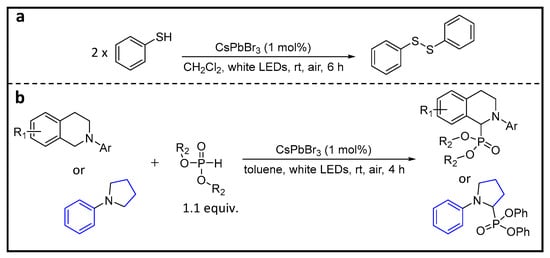
Wu et al. demonstrated the activation of a S–H bond using CsPbX
3 PNCs
[59].
Figure 16 shows the scheme of the oxidative coupling of thiophenol to form the disulfide, using CsPbBr
3 NCs under the conditions provided in the scheme. Notably, the choice of solvent plays a pivotal role in the reaction success. Changing from CH
2Cl
2 to CH
2Br
2/toluene/hexane significantly reduces the product yield, likely due to the conversion of the stable CsPbBr
3 to CsPbCl
3 (larger bandgap) and its relatively poor stability in various solvent systems. The most optimal solvent combination identified for this coupling reaction is CH
2Cl
2/cyclohexane and occurs with a 98% yield. In contrast, the reaction yields for CsPbI
3 and CsPbCl
3 are 58 and 12%, respectively. Furthermore, the versatility of this reaction extends to a wide range of substrates, including aromatic, aliphatic, and symmetric and unsymmetric compounds with electron-donating and -withdrawing groups. Remarkably, it consistently provides high yields. The authors proposed plausible mechanisms for this reaction. In this mechanism, thiol S and H coordinates with Pb and Br, respectively. Upon photoexcitation, electrons and holes transfer to H and S, respectively, eventually forming the corresponding radicals. As a result, two thiyl radicals combine to form disulfide, while two H radicals combine to generate H
2.
Figure 16. (a) Thiol-coupling reaction; (b) cross-dehydrogenative coupling reaction between tertiary amines and phosphite esters.
Moreover, researchers have also demonstrated a fascinating cross-dehydrogenative coupling reaction involving tertiary amines, targeting C–H bonds adjacent to amino nitrogen atoms and phosphite esters, as illustrated in Figure 16b.
7. Chiral Perovskite NCs
A material is considered chiral if its mirror image cannot be superimposed onto it. Chirality has been identified, for example, in carbohydrates and amino acids (except for glycine). Chiral catalysts have found extensive use in the asymmetric synthesis of pharmaceuticals. Recently, researchers have explored chiral MHPs to manipulate charge, light, and spin for applications in chiral optoelectronics, ferroelectrics, spin LEDs, spintronics, solar cells, and more
[60]. Hybrid perovskites offer more exciting properties compared to inorganic- and organic-based perovskites, including exceptional diffusion lengths and fewer defects. Additionally, introducing chiral organic ligands into the perovskite crystal structure can induce chirality. The transfer of chirality can take place through the establishment of chemical bonds or even via spatial interactions between a chiral and an achiral system.
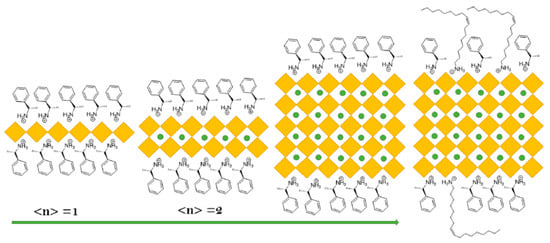
Reduced dimension perovskites can be represented by L2(APbBr3)n-1PbBr4 where L is surface organic chiral ligands, and A is a monovalent cation; n represents the number of inorganic layers between chiral organic ligands. Colloidal PNCs can be prepared with layers from n = 1 to ∞ by controlling the ration of L, A, and PbBr2, schematically shown in the Figure 17; L can be the R- or S-1-phenylethylammonium cation. Notably, the surface properties can be adjusted by the introduction of distinct chiral ligands.
Figure 17. Chiral perovskite NCs. NCs formulated with L2(APbBr3)n−1PbBr4. Right: proposed structure of NCs used in this study where n = ∞. Reproduced with permission. Copyright: 2023, American Chemical Society.
Production of N–C Axial Chiral N-Heterocycles Using Chiral CsPbBr3 NCs
Asymmetric catalysis requires the presence of a chiral-NC surface. Earlier research suggests a specific set of chiral ligands (ammonium-based) for perovskites. These chiral ligands are introduced to the perovskite either through direct synthesis or via post-synthetic ligand exchange. In the case of direct synthesis, chiral ligands impact the crystal structure, while post-synthesis incorporation leads to chirality due to the distortion of the NC surface. Recently, Yan et al. have demonstrated asymmetric photochemical synthesis using R-PEA hybridized perovskite PEA/CsPbBr
3 NCs as photocatalysts
[61]. These NCs were able to oxidize N-arylamine and produce N–C axially chiral N-heterocycles (N-arylindoles) under visible light. A range of R-PEA/CsPbBr
3 NCs were synthesized using emulsion synthesis, hot injection, or high-energy tip sonication methods. Among them, the tip sonication method exhibited the highest coverage of chiral ligands. The PNC surfaces were coated with both chiral ligands and non-chiral oleylammonium ligands, as depicted in
Figure 17.
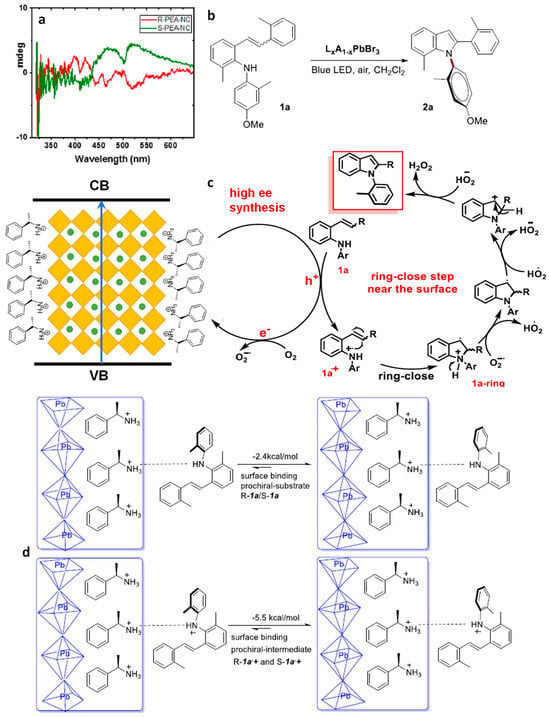
Typically, optically active compounds are assessed using circular dichroism (CD) spectroscopy. The chiral R- and S-forms of CsPbBr3 NCs displayed distinct CD signals at 515 nm and demonstrated anisotropy near the bandgap, as illustrated in Figure 18a. This CD response denotes the electronic interaction between the perovskite NC lattice and the chiral ligand. Chiral PEA/CsPbBr3 NCs were employed for the conversion of 1a to 2a (in Figure 18). Interestingly, a lower surface coverage with chiral ligands (9%, 17%, and 47%) yielded a higher enantiomeric excess (ee), specifically, 63%, 83%, and 99%, respectively. This suggests that a greater surface coverage enhances enantioselectivity.
Figure 18. (a) Circular dichroism (CD) spectrum of R-NC-3 and S-NC-3; (b) illustration of N–C axially chiral atroposelective N-heterocyclization using L2(APbBr3)n−1PbBr4 photocatalyst; (c) proposed mechanism for N-arylindole; (d) DFT studies, binding energy discrepancies between the chiral surface and substrate 1a (up), 1a+ (after hole transfer). Reproduced with permission. Copyright: 2023, American Chemical Society.
Figure 18c presents the proposed mechanism for the formation of highly enantioselective N-arylindoles; the substrate 1a approaches the chiral NC surface via the weak hydrogen bonding between the N-arylindol 2a and the enantiomerically pure PEA. After light excitation, the photogenerated holes transfer to 1a, resulting in 1a+, which undergoes ring closing on the surface-bound prochiral substrate (in Figure 18c).
Density functional theory (DFT) calculations were conducted to differentiate the preferred attachment of R- and S-1a substrates to the R-PEA chiral surface of CsPbBr3 NCs. These calculations revealed a −2.4 kcal/mol preferred binding energy of prochiral N-arylamine (1a) with R-configuration compared to its enantiomeric S-configuration counterpart, binding weakly through H-bonding to the R-PEA chiral surface (in Figure 18d). This suggests that the discrimination step occurs at the beginning of the catalytic cycle. Additionally, differences in surface binding energy after a photoinduced hole transfer indicated a −5.5 kcal/mol preferred binding energy of prochiral-R-1a+ over the prochiral-S-1a+ intermediate (in Figure 18d). Interestingly, during the ring-closing step, both R- and S-forms exhibited the same transition energy. Overall, the binding preference of R-PEA chiral surface of the CsPbBr3 NCs governs the N–C axial chirality of the N-heterocycles.
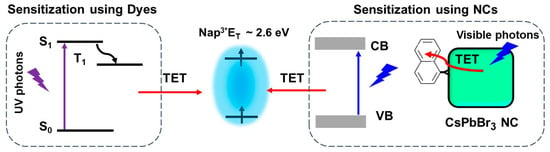
8. Triplet Energy Transfer from Dyes and NCs to Substrates
Polycyclic aromatic hydrocarbons (PAHs) are organic compounds extensively explored for their singlet and triplet states in various applications, including photocatalysis, solar energy, light emission, photon up-conversion, and room temperature phosphorescence
[62][63][64][65][66]. Nonetheless, the direct generation of their triplet state through optical excitation proves to be challenging due to their inherently dark nature and the inefficiency of intersystem crossing (ISC). Therefore, a sensitization approach has been adopted to generate PAH triplets, employing sensitizers, such as boron-dipyrromethane (BODIPY) dyes and fullerenes
[67][68][69]. These sensitizers absorb light to populate singlet states, which then efficiently undergo ISC to populate triplets. Subsequently, these triplets can transfer their energy to PAHs (acceptor) through Dexter-type triplet energy transfer (TET), as illustrated on the left side of
Figure 19. However, during the ISC process, there can be an intrinsic energy loss in the order of 0.5 eV or more, which leads to practical issues in visible-to-UV light up-conversion applications
[70].
Figure 19. (Left) Sensitization of naphthalene triplets (Nap3*) using dyes: light absorption populates the first singlet excited state (S1), which is followed by intersystem crossing (ISC) to generate the first triplet states (T1). These T1 states can subsequently transfer their energy to Nap via Dexter-type triplet energy transfer (TET) mechanism. Considering that the energy of Nap3* is ∼2.6 eV and that there is an estimated energy loss of ∼0.5 eV during the ISC process, it implies that the energy of S1 of the dye must be at least ∼3.1 eV, indicating that UV photons are required to drive the sensitization process; (Right) Sensitization of naphthalene triplets (Nap3*) using semiconductor CsPbBr3 NCs: light absorption leads to the generation of band edge excitonic states (X) within the NCs. These excitonic states can directly undergo TET to produce Nap3*. This means that visible photons are capable of efficiently driving the sensitization process.
Semiconductor NCs present significant benefits compared to molecular photosensitizers due to their easily achievable preparation, robust photostability, size-dependent electronic and photophysical characteristics, high molar extinction coefficients, and simple post-synthesis functionalization
[71]. Moreover, the minimal energy separation of 1 to 15 meV between bright and dark states, as opposed to the singlet–triplet splitting observed in organic molecules, renders NCs promising candidates for the sensitization process at the limit of energy conservation, driven by visible photons
[71][72]. Among the diverse arrays of NCs, PNCs have excellent optical properties, such as tuneable bandgaps in the UV–visible region, near unity PL QY, and potential as a triplet photosensitizer
[73]. These attributes position PNCs as promising candidates for generating triplet states in target substrates. On the right side of
Figure 19, an illustration shows the sensitization of naphthalene using confined CsPbBr
3 NCs, which possess a bandgap exceeding 2.6 eV, effectively aligning with the triplet energy (E
T) of approximately 2.6 eV
[74]. To achieve efficient TET from NCs to acceptors, the NC bandgap should be greater than the acceptor triplet energy level to be in resonance.
8.1. Demonstration of TET from Quantum Confined PNCs to Substrates
The utilization of visible light captured by long-lived triplet excited states has found widespread application in photocatalytic organic synthesis and photon up-conversion across various spectral ranges. An effective strategy involves storing the excitation energy of NCs in long-lived molecular triplet states through TET to PAHs. In this context, the combination of PNCs and PAHs allows for efficient TET, leveraging the strong light-harvesting capability of PNCs and surpassing the short emission lifetimes. This combination facilitates effective energy transfer, enabling the utilization of long-lived triplet states for desired applications in photon up-conversion and other related fields
[75]. For example, in a study conducted by Wu et al., the investigation focused on the size-dependent carrier density probability at the surface of CsPbBr
3 NC and its linear correlation with TET rates
[76]. They employed CsPbBr
3 NCs with varying edge lengths (L), ranging from 11.2 to 3.5 nm, resulting in bandgap variations from 2.43 to 2.73 eV. Triplet acceptor 1-pyrenecarboxylic acid (PCA) was introduced due to its triplet energy (E
T) of 2 eV and favourable band alignment with CsPbBr
3.
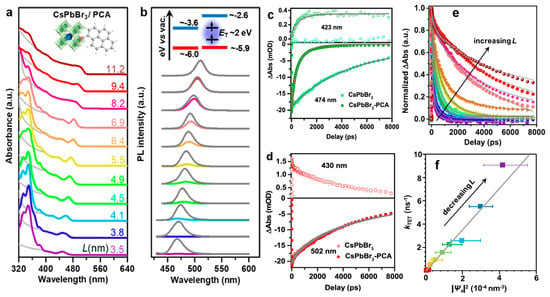
All CsPbBr3 NCs in the size range of 11.2 to 3.5 nm possessed bandgap energies higher than the ET of PCA, theoretically enabling TET. To explore size-dependent TET, PCA was grafted onto various-sized CsPbBr3 NCs via a carboxylic group. Figure 20a shows the absorption spectrum of the CsPbBr3-PCA complex (1:1 ratio of different-sized CsPbBr3 NCs and PCA), which was compared to that of pristine CsPbBr3 NCs, revealing a blue-shift in the lowest-energy absorption peak due to QCE as L decreased from 11.2 to 3.5 nm.
Figure 20. (a) Absorption spectra of CsPbBr3 NCs (grey solid lines) and NC–PCA complexes (coloured solid lines) with different NC sizes (edge length L indicated) dispersed in hexane. The variation between them is due to PCA absorption. The inset illustrates the schematic structures of NCs and PCAs; (b) PL spectra of CsPbBr3 NCs (grey solid lines) and NC–PCA complexes (coloured solid lines) excited at 420 nm. The inset illustrates the schematic energy level alignment between NCs and PCAs; (c) TA kinetics probed at the exciton bleaching (XB) centre (∼474 nm; solid symbols) and at the PCA triplet absorption (∼423 nm; open symbols) of L = 4.9 nm NCs (squares) and NC–PCA complexes (circles). The grey solid lines represent their fits using stretched-exponential functions; (d) Similar plot as in c for L = 9.4 nm NC–PCA complexes; (e) Normalized TA kinetics probed at the XB feature of NC–PCA complexes with varying NC sizes (coloured symbols). The grey solid lines represent their fits; (f) size-dependent TET rate (kTET) plotted as a function of carrier probability density at the NC surface (|Ψs|2) for different NC sizes (coloured symbols). The grey solid line depicts a linear fit. Reproduced with permission. Copyright: 2019, American Chemical Society.
In the analysis of the CsPbBr3-PCA complex, the absorption spectrum, depicted by the coloured line in Figure 20a, reflects features from both the CsPbBr3 NC and PCA (with PCA absorbing below the 400 nm region). To delve into the mechanism of TET from CsPbBr3 NCs to PCA molecules, steady-state PL for the CsPbBr3-PCA complex was systematically examined. Figure 20b showcases the steady-state PL spectra of CsPbBr3 NCs and the CsPbBr3-PCA complex under selective excitation of NCs at 420 nm.
Notably, the PL quenching efficiency witnessed a remarkable increase from 0.6% for bulk-like NCs with an L of 11.2 nm to an impressive 99% for the more strongly confined 3.5 nm-sized NCs. The energy level diagram presented in the schematic inset of Figure 20b elucidates that this substantial PL quenching can be attributed to the efficient TET process from the CsPbBr3 NCs to the PCA molecule. This compelling finding underscores the efficacy of TET, particularly in the context of strongly confined CsPbBr3 NCs.
Transient absorption spectroscopy (TAS) was employed to monitor TET dynamics. Figure 20c show the kinetics of exciton bleaching (XB) in free NCs and NC–PCA complexes. This indicated a significant shortening of the XB lifetime in NC–PCA complexes, accompanied by the appearance of an absorption feature at ~ 423 nm corresponding to the T1 → Tn transition of PCA triplets. This provided clear evidence of ultrafast TET from NCs to PCAs.
The ultrafast quenching of NC excitons is conclusively attributed to TET from NCs to PCAs. Intriguingly, this ultrafast TET stands in stark contrast to the negligible TET observed in 9.4 nm bulklike NCs, as depicted in Figure 20d. The XB lifetimes in both free NCs and NC–PCA complexes remained similar, and no discernible triplet absorption feature was identified (Figure 20d). This striking disparity in behaviour between small- and large-sized NCs underscores the pivotal role played by the QCE in facilitating efficient TET.
Figure 20e presents the normalized kinetics of the exciton bleaching feature (XB) for various CsPbBr3-PCA complexes utilized in the study. Notably, the TET rate demonstrates a clear trend increasing with the decreasing size of NCs. Particularly, the strongly confined 3.5 nm NCs exhibit a notably higher TET rate. The QCE primarily influences the energetics (driving force and spectral overlap) and electronic coupling involved in the TET process, particularly if TET follows the well-established Dexter mechanism. To unravel the effect of the driving force and spectral overlap, the TET was examined using similar-sized NCs with varying emission peaks achieved by altering x in the CsPbBr3−xClx NCs. Intriguingly, NC–PCA complexes with different x values displayed remarkably similar XB recovery kinetics, suggesting that their TET kinetics should also align. This implies that spectral overlap and driving force play negligible roles in the TET for this system. Consequently, the inference is that quantum confinement primarily influences TET through electronic coupling.
Smaller NCs with stronger QCE are known to possess higher carrier probability densities at their surfaces, leading to stronger electronic coupling with attached acceptors. To understand the size-dependent TET rate (kTET), the carrier probability density at the CsPbBr3 NC surface denoted as |Ψs|2 was plotted against kTET, revealing an increase from nearly 0 for 11.2 nm-sized NCs to 9.1 ± 0.1 ns−1 for 3.5 nm-sized NCs. The linear fitting of this data suggests that the wave function exchange between CsPbBr3 NCs and PCA molecules is the dominant factor governing this Dexter-type mechanism. Notably, the absence of a hole transfer pathway from NCs to PCA in this study further underscores the impact of QCE on efficiently mediating TET to PCA molecules with decreasing NC size.
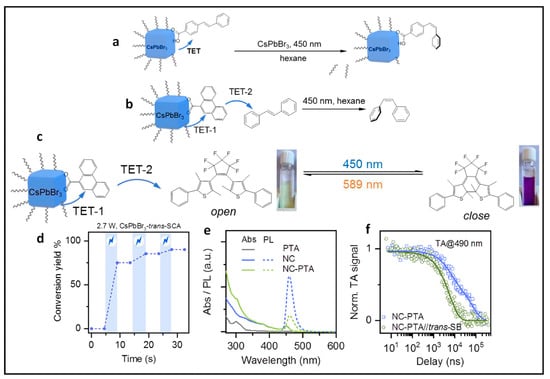
8.2. Isomerisation of Surface-Anchored Stilbene
Wu et al. demonstrated visible light-driven molecular isomerization and cycloaddition reactions via the TET approach from CsPbBr
3 and CsPbBr
3−xI
x PNCs to the substrates
[77]. To showcase photoisomerization, the authors selected trans-4-stilbenecarboxylic acid (trans-SCA), whose carboxylic group anchors to the of PNC surface. The stilbene triplet energy (E
T) is ~2.13 eV for the trans isomer and ~2.47 eV for the cis isomer. Consequently, 3.5 nm-sized CsPbBr
3 NCs with an excitonic peak at approximately 455 nm (2.73 eV) were chosen to drive the reaction via TET.
Figure 21a illustrates the isomerization of trans SCA to cis SCA in hexane under 5 mW 450 nm illumination. The conversion yield (CY) was calculated based on the absorption coefficients of trans- and
cis-stilbene. An illumination power of 2.7 W enabled the reaction to reach the steady state within 15 s. The light-modulated CY is depicted in
Figure 21d, showing an increase in CY under illumination, ultimately reaching 90.1%. Furthermore, when photoisomerization was conducted with CsPbBr
3−xI
x NCs, whose bandgap slightly exceeded the ET of
trans-SCA (approximately 2.25 eV), it was intriguingly transformed to cis-SCA at a slower rate than that of CsPbBr
3 NCs.
Figure 21. (a) Photoisomerization of surface tethered stilbene; (b) the 9-phenanthrene carboxylic acid (PTA) ligands are anchored on CsPbBr3 NCs surfaces, whereas the stilbene molecules are freely diffusing in solution; (c) photoisomerization of diarylethene; (d) light-modulated conversion yields under 2.7 W 450 nm illumination, the blue shaded areas correspond with light-on; (e) absorption (solid lines) and PL (dashed lines) spectra of CsPbBr3 NCs with (green) and without (blue) surface-anchored PTA ligands; (f) nanosecond-TA kinetics probed for NC-sensitized 3PTA* with (green open circles) and without (blue open squares) trans-stilbene in the solution. Reproduced with permission. Copyright: 2022, John Wiley and Sons.
8.3. Isomerization of Stilbene in Solution Using Relay
Isomerization of organic molecules in a free form, i.e., non-anchored onto the NC surface, was also possible using surface-anchored triplet transmitters, which act as a relay to accept energy from photoexcited NCs and pass it to the annihilator in solution. Wu et al. demonstrated that 9-phenanthrene carboxylic acid (PTA) with ET of ~2.64 eV as the transmitter ligands showed the TET from 3.5 nm-sized CsPbBr3 NCs to trans-stilbene as shown in Figure 21b. Figure 21e shows the absorption and PL spectra of CsPbBr3 NCs as well as those of PTA-anchored CsPbBr3 NCs. A significant PL quenching (69.2%) was observed in the presence of PTA ligands. In Figure 21f, TA kinetics show that the average lifetime (~42 µs) of CsPbBr3 NC sensitized 3PTA* shortened to ~5.8 µs for the complex of NC-PTA and trans-stilbene. This indicates an efficient TET-2 (86.1%) from 3PTA* to stilbene. This reaction was performed under 2.7 W 450 nm illumination; the CY was of 82.7% in 30 s. Thus, TET and photoisomerization can be comparable between surface-tethered and relayed systems.
Additionally, authors used the relay method to demonstrate the ring-closing isomerization of diarylethene (DAE), as shown in Figure 21c. The DAE open form can be converted into the DAE closed form via efficient TET-2 from the 3PTA* to DAE open form. This could be clearly seen since the colour changed from colourless to purple, corresponding to their absorption at of 270 nm and 565 nm of DAE open and closed forms, respectively. However, control experiments failed to convert the open-to-closed form with direct 450 nm laser illumination. Notably, the selective excitation of the DAE closed form with a 589 nm laser induced the conversion to the open form. This reversible photochromic reaction finds utility in applications such as light-induced information coding and patterning.
The relay method can also be extended to intermolecular [2 + 2] cycloaddition reactions of acenaphthylene (ACN) to dimerization. The ET of ACN was in the range of 1.87–1.95 eV, which is lower than that of PTA ligands.
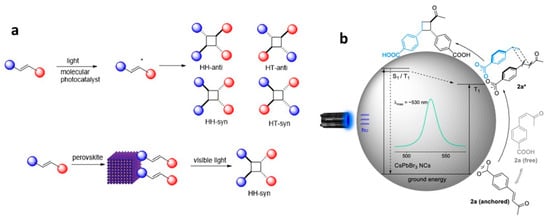
8.4. Photocatalytic 2 + 2 Cycloadditions
The direct utilization of long-lived triplet states using molecular photocatalysts for organic molecule activation has been extensively explored. This often entails the TET from these molecular photocatalysts to organic substrates, inducing non-selective 2 + 2 cycloaddition reactions, as depicted in
Figure 22a. In contrast, using PNC surfaces as templates, dynamically anchored substrates facilitated efficient TET to the substrate and achieved regioselective head-to-head (HH) 2 + 2 cycloaddition reactions in a
syn-mode. This
syn-binding mode promotes the formation of thermodynamically unfavourable 2 + 2
syn-cyclobutane products, a feat beyond the realm of molecular photocatalysis. Yan et al. demonstrated the photocatalytic synthesis of highly diastereomeric
syn-selective 2 + 2 olefin cycloadditions using bulk-like PNCs as photosensitizers via TET
[78].
Figure 22b exemplifies the photocatalytic 2 + 2
syn-cycloaddition reaction for substrate 2a (in
Figure 22b) under optimized conditions, illustrated in the
Figure 22b.
Figure 22. (a) Molecule- and perovskite-induced photocatalysis for TET based 2 + 2 cycloaddition. HH: head-to-head, HT: head-to-tail; star represents the excitation of the molecular photocatalyst; (b) proposed mechanism for the intermolecular 2 + 2 cycloaddition facilitated by CsPbBr3 NCs through TET induced by visible light. Reproduced with permission. Copyright: 2022, American Chemical Society.
The anchoring of the substrate to the PNC surface through a carboxylic group along with its lower triplet energy than that of bulk-like PNCs mediated the TET. Comparative studies revealed that the same reaction with an Ir(ppy)3 molecular photocatalyst yields a mixture of products, with HH-anti and HH-syn as major and minor products, respectively, with trace amounts of HT-anti and HT-syn products, where the triplet energy acceptor remains in a free (unbound) state. Moreover, the applicability of photocatalytic 2 + 2 cycloadditions can be broadened to encompass both homo- and hetero-variations involving other substrates possessing triplet energies lower than those of PNCs.
This entry is adapted from the peer-reviewed paper 10.3390/nano14010094