Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Alzheimer’s disease (AD) is the most common cause of dementia in the elderly and its development is linked to multifactorial interactions between the environment, genetics, aging and lifestyle. The pathological hallmarks in AD are the accumulation of β-amyloid peptide (Aβ), the hyperphosphorylation of tau protein, neurotoxic events and impaired glucose metabolism. Due to pharmacological limitations and in view of the prevailing glycemic hypometabolism, the ketogenic diet (KD) emerges as a promising non-pharmacological possibility for managing AD, an approach that has already demonstrated efficacy in addressing other disorders, notably epilepsy. The KD consists of a food regimen in which carbohydrate intake is discouraged at the expense of increased lipid consumption, inducing metabolic ketosis whereby the main source of energy becomes ketone bodies instead of glucose. Thus, under these dietary conditions, neuronal death via lack of energy would be decreased, inasmuch as the metabolism of lipids is not impaired in AD. In this way, the clinical picture of patients with AD would potentially improve via the slowing down of symptoms and delaying of the progression of the disease.
- Alzheimer’s disease
- ketogenic diet
- ketone bodies
- neurodegenerative diseases
1. Epidemiology, Neuropathological Insights and Symptoms in Alzheimer ’s Disease
Alzheimer’s disease (AD) is the most prevalent form of dementia [1][2][3][4]. It is also considered to be a growing burden for the health care system and is one of the most expensive chronic diseases of old age [3][5][6][7]. In 2010, a total of 604 billion USD was spent on AD globally; these direct medical costs are exacerbated by lost productivity attributed to this debilitating disease. Projections for the year 2050 estimate that such costs would surpass 1 trillion USD in total [6][8][9][10]. These economic expenses are paralleled with the incidence of patients with Alzheimer’s, several of which are forecasted to rise above 150 million by 2050 [10][11]. In addition, AD has an intense impact on the individual and social spheres, with caregivers of affected people being more vulnerable to anxiety and depression [5][12]. Therefore, the functional capacity and autonomy of the elderly may be more important than mortality, as they relate to quality of life [6]. In all populations, AD is prevalent at advanced ages, notably between 70 and 80 years [2][13]. However, differences were found in the association of AD among ethnic groups and sex [14][15]. Importantly, a greater susceptibility to developing AD has been reported in Caucasians, Japanese, and women, as nearly two-thirds of people diagnosed are women [16][17].
Neuropathologically, AD is characterized by the deposition of β-amyloid peptides (Aβ) and the accumulation of neurofibrillary tangles (NFTs) composed of hyper-phosphorylated tau protein. Under physiological conditions, tau protein exists as a soluble and unfolded protein that interacts with tubulin to promote the assembly and stabilization of microtubules [18][19]. Aβ peptides can self-aggregate into soluble oligomers, or into insoluble fibers, forming the amyloid plaques. The Aβ from 36 to 43 amino acids is a product of proteolytic processing of protein amyloid precursor (APP), a type I transmembrane protein [20]. The cleavage of APP by the enzyme β-secretase (BACE-1) generates a C-terminal fragment (β-CTF), which is an immediate substrate for the γ-secretase enzyme that cleaves β-CTF to produce a spectrum of Aβ peptides with different lengths, i.e., the amyloidogenic pathway [21]. Conversely, when APP is cleaved by the α-secretase enzyme, it generates a C-terminus that subsequently undergoes cleavage by γ-secretase. This pathway, known as the non-amyloidogenic pathway, leads to the production of a shorter APP fragment (p3). This seemingly innocuous fragment may, in fact, hold pivotal roles in various processes, including cell growth, adhesion, synaptic plasticity, and the regulation of metal ion homeostasis [22][23].
Moreover, the amyloid hypothesis postulates that the accumulation of Aβ leads to a neurodegenerative cascade, resulting in synaptic dysfunction, NFT formation and, ultimately, neuronal loss in susceptible brain regions. Thus, AD is associated with neuronal degeneration, manifesting selective impairments in the hippocampus and neocortex regions, predominantly affecting cholinergic neuronal populations, and resulting in neurological impairments [24][25][26]. While the manifestation of symptoms may exhibit heterogeneity, AD is characterized by a progressive and irreversible decline in memory, cognition, thinking and language [27][28][29]. This decline culminates in diminished independence in performing daily tasks, accompanied by behavioral and motor impairments, as well as escalating psychiatric symptoms. These challenges tend to intensify as the neurodegenerative syndrome progresses [16][24][30]. Moreover, shared mechanisms of cell death in AD are related to increased oxidative stress, mitochondrial injury, hypometabolism, disruption of the blood–brain barrier (BBB) and neuroinflammation [31][32][33]. In the latter process, the increased formation of Aβ and NFTs is associated with the presence of activated microglia, cytokines, cyclooxygenase 2 (COX-2) and other inflammatory substances [28][32].
2. Risk Factors for AD
Despite 115 years having passed since Alois Alzheimer first described the disease, our understanding of AD remains incomplete. Consequently, researchers have explored genetic and environmental factors as potentially influential elements in the development of this condition [34][35]. Genetic risk factors for AD include mutations in amyloid precursor protein (APP), presenilin 1 (PSEN1), presenilin 2 (PSEN2) and apolipoprotein E4 (APOE4). Nonetheless, APOE4 stands as the most extensively established genetic risk factor for AD susceptibility beyond the age of 65 years [36][37][38][39]. It is linked to an elevated risk of NFT formation and a reduction in Aβ clearance, resulting in the accumulation of neurotoxic fragments [37][38]. Furthermore, while genes play a role in the risk of developing AD, they may account for only a modest portion of that risk [2][36]. It is important to highlight that the risk factors identified for AD share features and are not independent [40]. Notably, some of these factors encompass treatable medical conditions like stroke, hypertension and diabetes [36]. Epidemiologic studies revealed that the risk of AD is increased by 50–100% by type 2 diabetes mellitus (T2DM). A notable observation is that the APOE4 gene serves as a risk factor for the development of type 2 diabetes mellitus (T2DM), although it is important to recognize that not all individuals with T2DM possess the APOE4 gene. Additionally, this apolipoprotein has been linked to a reduction in glucose consumption by the brain [41]. Thus, T2DM is linked to an increased risk of developing late-onset AD (LOAD) [42][43]. Regarding modifiable environmental risks, numerous reviews have emphasized the substantial evidence associating smoking [42], nutrition [44] and obesity [45], and such findings demonstrate that behavioral aspects can influence the onset of clinical manifestations of AD [46][47][48].
3. Pharmacological Treatment for AD
Currently, there is neither a cure for AD [49] nor an effective drug for prevention or treatment that modifies the progression of the disease [41][50]. Given the severity of AD, advances in pharmacological treatments are progressing slowly [51], as only a few drugs are approved. The drug therapy for AD is symptomatic or palliative and not effective for advanced stages of the disease [49][52][53][54]. Such pharmacological treatments are focused on improving cholinergic transmission, and they are divided by the mechanism of action into two classes: cholinesterase inhibitors (ChEIs), used for mild to moderate stages, and an NMDA receptor antagonist (N-Methyl-D-aspartate), Memantine [49], used for moderate to severe stages or in cases of intolerance and contraindication [8][55][56]. ChEIs have moderate symptomatic benefits regarding cognition, functionality and behavior [36][57][58]. This class includes Donepezil, Galantamine and Rivastigmine [59]. Nevertheless, these drugs exhibit variations in certain pharmacological characteristics: Donepezil and Rivastigmine boast longer half-lives, but, in addition to acetylcholinesterase inhibition, Rivastigmine also deactivates butyrylcholinesterase. On the other hand, Galantamine demonstrates an additional enhancement in nicotinic receptor transmission [60][61]. As for the NMDA receptor antagonist, Memantine improves cognition, functionality and the management of agitation and aggression [36][58][62]. Furthermore, combining Memantine with Donepezil can lead to improved patient outcomes [61].
Additional treatment options are non-pharmacological, often more cost-effective and dependent on human effort [63]. This category encompasses numerous suggested approaches, including socialization [64], cognitive training [65], calorie restriction and exercise [8][60]. Among these alternatives, the ketogenic diet (KD) is currently under investigation as an adjuvant therapy [66][67][68][69].
4. Ketogenic Diet and Ketone Body Biosynthesis
The KD is a diet based on reducing carbohydrate consumption and increasing lipid intake [70][71]. This leads to a decrease in the use of glucose, which is no longer the main energy source, promoting the use of ketone bodies (ketones) from the breakdown of fatty acids (FAs) [72][73][74]. The hepatic metabolism of FAs produces ketones commonly used as substrates for energy: acetoacetate, acetone and beta-hydroxybutyrate (BOHb) [75][76]. Normally, ketones are produced in starvation [77], fasting [78][79], prolonged physical exercises [80], pregnancy [81] and in diets with high-fat and low-carbohydrate rates [82]. Thus, when glucose stores in the body are low, more FAs are made available to the liver for oxidation, leading to the consequent production of energy-rich molecules, mainly acetyl-CoA. Acetyl-CoA can enter the citric acid cycle in the liver or be used for the synthesis of ketones. Once in the liver cells, the fatty acid will be directed to the mitochondrial matrix by carnitine palmitoyltransferase (CPT1/2) where it initially undergoes a β-oxidation generating Acetyl-CoA that will undergo the process of ketogenesis [83]. In sequence in the process, the thiolase-2 enzyme acts in the conversion of two molecules of Acetyl-CoA to Acetoacetyl-CoA (AcAc-CoA) [83][84]. This molecule undergoes catalysis by the enzyme 3-Hydroxymethyl glutaryl-CoA synthase 2 (HMGCS2), resulting in the generation of hydroxymethylglutaryl-CoA (HMG-CoA) [84]. Subsequently, HMG-CoA is converted into acetoacetate and Acetyl-CoA via the catalytic action of HMG-CoA lyase [85]. Furthermore, acetoacetate can be reduced to D-β-hydroxybutyrate (D-βOHB) or decarboxylated to acetone [83]. After their formation, ketones are released from cells by monocarboxylate transporters (MCT1/2) and fall into the bloodstream to reach extrahepatic tissues for terminal oxidation [83]. Through the same MCT1/2 channels, ketone bodies enter the mitochondrial matrix of cells where they undergo the action of Succinyl-CoA: 3-ketoacid CoA transferase (SCOT) that transfers the CoA portion of succinyl-CoA to form Acetoacetyl-CoA [86]. The final part of the inverse process leads to the formation of Acetyl-CoA that is introduced into the TCA cycle for the formation of ATP that is used as an energy source in cases of glucose deprivation [87] (Figure 1).
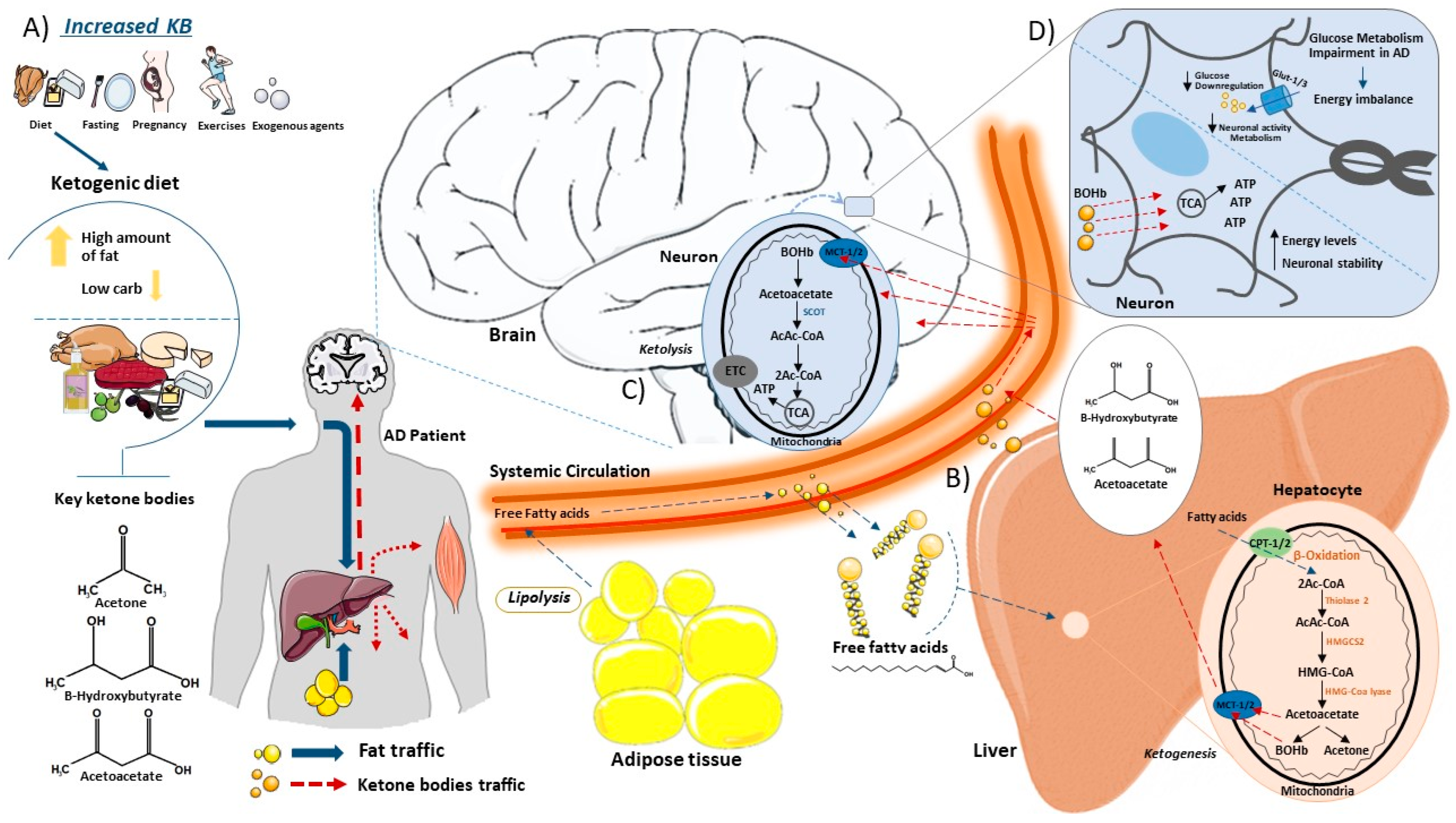
Figure 1. This schematic illustrates the synthesis and catabolism of ketone bodies within the body. (A) Ketones are produced in the liver under certain conditions. Consuming a high-fat low-carbohydrate diet promotes the production of ketone bodies as acetone, beta-hydroxybutyrate (BOHb) and acetoacetate in the liver. (B) Once in the bloodstream, free fatty acids from the ketogenic diet or adipocytes (lipolysis) enter the liver via the hepatic portal vein, where they participate in the ketogenesis process. Inside liver cells, free fatty acids access mitochondria via Carnitine Palmitoyltransferase (CPT) transporter channels. Within the mitochondria, fatty acids undergo oxidation, leading to the formation of essential ketone bodies. These ketone bodies exit the mitochondria through Monocarboxylate Transporter (MCT) channels, eventually leaving hepatocytes and entering the bloodstream, en route to the brain. (C) In the brain, ketone bodies enter the neuronal mitochondrial matrix through Monocarboxylate Transporters (MCT channels), where they undergo ketolysis. The action of Succinyl-CoA on ketone bodies promotes the formation of Acetyl-CoA, which subsequently integrates into the tricarboxylic acid (TCA) cycle for ATP generation. (D) Alzheimer’s disease is intricately linked to impaired glucose metabolism, resulting in an energy imbalance. Reduced glucose levels in cells compromise neuronal activity and metabolism. In this scenario, with diminished glucose availability, ketone bodies become the primary source for ATP production, allowing cells to maintain high energy levels and neuronal stability. This figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
5. Types of KD
There are different types of KDs, listed as: classic long-chain triglyceride KD (LCT), medium-chain triglyceride KD (MCT), modified Atkins diet (MAD) and low glycemic index diet (LOGI) [66][88][89][90]. The four diets have the same original formula, characterized by a high rate of fat and low amount of carbohydrate in their composition. However, they have occasional variations in the composition weight and ingredient restrictions [90]. LCT offers around 90% of energy in the form of fat and 10% of carbohydrates and proteins [90]. The most recommended ratio is 4:1 to 3:1 (fats: proteins and carbohydrates), but the use of each diet can be evaluated based on the patient’s profile and the most appropriate type of diet [90]. The diet ratio represents the balance between fat and protein plus carbohydrate grams. For instance, a “1800 kcal 4:1 ratio classic KD” contains four times the grams of fat compared to protein. This ratio can be customized to enhance seizure management or to make it more accommodating for improved tolerance. In contrast to the standard KD, the MCTKD is not influenced by food ratios; instead, it depends on the proportion of calories derived from MCT oil as a crucial source of ketones [91].
MCT has a distinct composition, primarily comprising about 60% octanoic acid, an eight-carbon fatty acid, and roughly 40% decanoic acid, a ten-carbon fatty acid [92]. Unlike the conventional KD, which relies more on medium-chain fats for dietary energy, the MCT-based diet permits a broader inclusion of carbohydrates. Due to the swift metabolism of these shorter fatty acids, this distinction results in a more efficient synthesis of ketones [92]. The process starts with dietary triglycerides in the form of MCT supplements, undergoing breakdown in the gastrointestinal tract by specialized lipases with a preference for hydrolyzing medium-chain esters rather than long-chain esters. Consequently, MCTs are converted into medium-chain fatty acids, characterized by their carbon atom content ranging from six to twelve [93]. This unique property enables direct absorption through the intestinal wall, leading to swift transportation to the liver. Once in the liver, the medium-chain fatty acids, including decanoic acid and octanoic acid, undergo rapid metabolism via a process known as β-oxidation [93]. The integration of an MCT-rich diet with an elevated carbohydrate intake sets this approach apart from the conventional KD, offering a balanced and efficient means of achieving ketosis while harnessing the advantages of medium-chain fats [92].
Due to the highly restrictive dietary regimen and concerning side effects associated with KDs, their implementation in pediatric patients is challenging. In this context, the modified Atkins diet (MAD) emerges as a more balanced and easily applicable alternative dietary therapy. The advantages of increased tolerance and sustainable treatment approach could potentially position MAD as the preferred choice. Unlike the classic KD, MAD shares similar food choices but eliminates the need for precise ingredient weighing. It also deviates from a strict ketogenic ratio and lacks restrictions on protein, fluid and calories. MAD is used to treat some metabolic paroxysmal movement disorders, such as those observed in glucose transporter type 1 deficiency syndrome (GLUT1DS) [88][89][90].
Another type of diet that combines the principles of the Mediterranean diet with the macronutrient composition of a KD is the Mediterranean Ketogenic Diet (MKD). Inspired by the heart-healthy Mediterranean diet, the MKD incorporates high consumption of fruits, vegetables, whole grains and healthy fats, notably olive oil. Simultaneously, it aligns with the principles of a KD by emphasizing low carbohydrate intake and promoting a state of ketosis. The MKD includes the use of olive oil as a primary source of fat, moderate consumption of fish and poultry, and limited intake of red meat and processed foods. The MKD encompasses several common variations, including the Very Low-Calorie Ketogenic Diet (VLCKD) which is characterized by a low carbohydrate content (<50 g/day), 1–1.5 g of protein/kg of ideal body weight, 15–30 g of fat/day and a daily intake of about 500–800 calories [94]. In addition, the High-Fat Ketogenic Diet (HFKD) is based on a higher proportion of daily calories sourced from fat, typically ranging between 75–80%. In contrast to the traditional KD, the HFKD allows for a modestly increased protein intake, contributing to a more flexible nutritional profile [94].
6. Possible Risks of KD
The benefits of ketones produced in the liver go beyond the energy supply of tissues such as the brain, skeletal muscle and heart [83]. Ketones antagonize inflammatory processes and oxidative stress [95][96], acting as signaling mediators [83]. Although the KD presents possible benefits to the organism [97], its use can also promote adverse effects such as headache, gastrointestinal pain, constipation, nausea, fatty diarrhea, fatigue, vomiting and other gastrointestinal problems [98][99][100]. These symptoms are usually associated with acute use of the KD as reported in studies with young and adult patients [99][100]. All the symptoms caused by the KD in the first few days are usually called “keto flu” [100]. It has been shown that the acute symptoms pass after a short period of time and patients who use the KD for more than one year can report different types of symptoms, as vitamin and mineral deficiencies, kidney stones, hyperuricemia, lethargy and infectious diseases, which can be harmful [99].
7. The Use of the KD in ND
Since the 1920s, the KD has been employed as a therapeutic approach for various neurological disorders, most notably in the management of drug-resistant refractory childhood epilepsy [92][101][102]. More recently, its potential role has been explored in the context of several neurodegenerative diseases, including AD [66] and PD [102][103], as well as in psychiatric diseases such as schizophrenia [104][105], depression [106][107] and Borderline Syndrome [108]. The success behind the use of the KD is linked to its action in common features shared by most CNS diseases, such as glucose hypometabolism, energetic deficits, imbalanced GABA and glutamate transmission, inflammation and oxidative stress [70][92]. Notably in some neurodegenerative diseases, patients present failures in the expression of glucose receptors GLUT1 and GLUT3, which may lead to glucose hypometabolism, contributing to the aggravation of diseases [92][109][110]. For instance, Glut1 deficiency syndrome (Glut1DS) causes a delay in glucose transport through the blood–brain barrier (BBB), leading to decreased cerebrospinal fluid glucose levels even in the presence of normal blood glucose [111]. In this context, the KD can enhance the provision of ketones at the expense of carbohydrates, potentially leading to an increased production of ATP, protecting neurons from energetic deficits. This is supported by a study that demonstrated a 22% increase in the expression of BOHb transporters, a key component of the KD, in patients with schizophrenia [110]. Moreover, the imbalance between GABA and glutamate represents a common feature contributing to the characteristic seizures observed in many neuropsychiatric disorders [70]. Notably, the Medium-Chain Triglyceride Ketogenic Diet (MCT KD) achieves seizure control via decanoic acid, which selectively inhibits AMPA receptors as demonstrated in preclinical models [31][92]. Furthermore, the anti-inflammatory properties of the KD are associated with its ability to shift microglial cells from a pro-inflammatory state to an anti-inflammatory state, offering promise in the management of mental illnesses [112]. Additionally, the KD, primarily via its ketone constituents, exerts control over oxidative stress by influencing various metabolic and signaling factors [70][92]. This multifaceted action underscores the broad spectrum of the KD’s therapeutic potential in treating various CNS diseases, thereby encouraging further exploration of its utility in conditions such as AD.
8. The Rationality of KD Use in AD
The progressive deposition of Aβ peptides and increased levels of hyperphosphorylated tau protein trigger neurodegeneration and impaired glucose metabolism [113]. Indeed, deficiencies of the GLUT1 receptor have been reported in AD, leading to impairment of glucose transport through the blood–brain barrier (BBB), causing hypometabolism of glucose in the CNS and consequently, starvation of neurons because of inefficient glycolysis [92][109][114]. Thus, an adjuvant therapy route to overcome the energy inefficiency of glucose in AD may act as an alternative fuel for brain metabolism [115][116]. In this case, the KD offers high levels of fat that, when metabolized in the liver, generate ketones which can supply the brain energetically and prevent neuronal death and synapse loss [41]. KD treatment may lead to neuroprotective effects by reducing Aβ damage to mitochondria, increasing ATP production, and reducing oxidative stress and glutamate toxicity [117]. Improvements in mitochondrial function have been attributed to biochemical changes resulting from the inhibition of glycolysis and increased KB formation [41]. Furthermore, KD treatment elevates ketone levels, and this increase exerts a neuroprotective effect on aging brain cells by diminishing the expression of inflammatory and apoptotic mediators [41][118][119].
Indeed, intriguing hypotheses propose a multifaceted impact of ketones on gene expression alterations and the modulation of cell signaling cascades. These processes, in turn, appear to regulate neuronal excitability, bolster antioxidant defenses and maintain the redox balance within cells [120][121]. In addition, there are suggestions that KD treatment may impact the deposition of Aβ or tau, slowing down the underlying disease process [92][122]. Moreover, the KD may act to reestablish the misbalance between the GABAergic and glutamatergic neurotransmission present in AD [123][124]. It has been reported that Aβ may increase AMPA currents, leading to glutamatergic hyperactivity, neurotoxicity and memory loss in AD [125]. In addition, the KD showed improvement in neuronal survival via the inhibition of AMPA receptors [31][92]. Another crucial aspect is that KD therapy contributes to the preservation of synaptic activity. This is achieved by enhancing the GABA/Glutamate ratio via elevated levels of Krebs cycle intermediates and by activating ATP-sensitive potassium channels via mitochondrial metabolism [126]. Hence, the implementation of the KD may attenuate neuronal hyperexcitability that has been described in preclinical and early clinical stages of AD [127][128].
Also, neuroinflammation, brain cell atrophy and apoptosis observed in AD may be related to the deleterious cellular effects of phosphate toxicity. It seems that hyperphosphorylated tau engages in the significant utilization of phosphate; consequently, a diminished dietary phosphorus intake emerges as a potential strategy to mitigate the risk of AD [46]. Interestingly, some studies suggest that the KD may increase the excretion of phosphorus in urine. This could potentially lead to lower phosphate levels in the body [46].
This entry is adapted from the peer-reviewed paper 10.3390/metabo14010025
References
- Takada, L.T.; Caramelli, P.; Radanovic, M.; Anghinah, R.; Hartmann, A.P.; Guariglia, C.C.; Bahia, V.S.; Nitrini, R. Prevalence of potentially reversible dementias in a dementia outpatient clinic of a tertiary university-affiliated hospital in Brazil. Arq. Neuropsiquiatr. 2003, 61, 925–929.
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152.
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Alzheimer’s Disease International. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117.
- Schneider, J.A.; Arvanitakis, Z.; Leurgans, S.E.; Bennett, D.A. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann. Neurol. 2009, 66, 200–208.
- Qiu, C.; Kivipelto, M.; Von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128.
- Gutierrez, B.A.; Silva, H.S.; Guimarães, C.; Campino, A.C. Impacto econômico da doença de Alzheimer no Brasil: É possível melhorar a assistência e reduzir custos? Economic impact of Alzheimer’s Disease in Brazil: Is it possible to improve care and minimize costs? Cien. Saude Colet. 2014, 19, 4479–4486.
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33.
- Corbett, A.; Pickett, J.; Burns, A.; Corcoran, J.; Dunnett, S.B.; Edison, P.; Hagan, J.J.; Holmes, C.; Jones, E.; Katona, C.; et al. Drug repositioning for Alzheimer’s disease. Nat. Rev. Drug Discov. 2012, 11, 833–846.
- Stefanacci, R.G. The costs of Alzheimer’s disease and the value of effective therapies. Am. J. Manag. Care 2011, 17, S356–S362.
- Wong, W. Economic burden of Alzheimer disease and managed care considerations. Am. J. Manag. Care. 2020, 26, S177–S183.
- Vogt, A.S.; Jennings, G.T.; Mohsen, M.O.; Vogel, M.; Bachmann, M.F. Alzheimer’s Disease: A Brief History of Immunotherapies Targeting Amyloid β. Int. J. Mol. Sci. 2023, 24, 3895.
- Rao, A.K.; Chou, A.; Bursley, B.; Smulofsky, J.; Jezequel, J. Systematic review of the effects of exercise on activities of daily living in people with Alzheimer’s disease. Am. J. Occup. Ther. 2014, 68, 50–56.
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783.
- Chen, H.Y.; Panegyres, P.K. The Role of Ethnicity in Alzheimer’s Disease: Findings From The C-PATH Online Data Repository. J. Alzheimers Dis. 2016, 51, 515–523.
- Viña, J.; Lloret, A. Why women have more Alzheimer’s disease than men: Gender and mitochondrial toxicity of amyloid-beta peptide. J. Alzheimers Dis. 2010, 20, S527–S533.
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; Van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356.
- Canevelli, M.; Quarata, F.; Remiddi, F.; Lucchini, F.; Lacorte, E.; Vanacore, N.; Bruno, G.; Cesari, M. Sex and gender differences in the treatment of Alzheimer’s disease: A systematic review of randomized controlled trials. Pharmacol. Res. 2017, 115, 218–223.
- Selkoe, D.J. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends. Cell Biol. 1998, 8, 447–453.
- Spillantini, M.G.; Goedert, M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998, 21, 428–433.
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766.
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356.
- Turner, P.R.; O’Connor, K.; Tate, W.P.; Abraham, W.C. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol. 2003, 70, 1–32.
- Perez, R.G.; Zheng, H.; Van der Ploeg, L.H.; Koo, E.H. The beta-amyloid precursor protein of Alzheimer’s disease enhances neuron viability and modulates neuronal polarity. J. Neurosci. 1997, 17, 9407–9414.
- Gauthier, S.; Wirth, Y.; Möbius, H.J. Effects of memantine on behavioural symptoms in Alzheimer’s disease patients: An analysis of the Neuropsychiatric Inventory (NPI) data of two randomised, controlled studies. Int. J Geriatr. Psychiatry 2005, 20, 459–464.
- Tarawneh, R.; Holtzman, D.M. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb. Perspect. Med. 2012, 2, a006148.
- Iqbal, K.; Wang, X.; Blanchard, J.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Alzheimer’s disease neurofibrillary degeneration: Pivotal and multifactorial. Biochem. Soc. Trans. 2010, 38, 962–966.
- de la Rubia Ortí, J.E.; Fernández, D.; Platero, F.; García-Pardo, M.P. Can Ketogenic Diet Improve Alzheimer’s Disease? Association With Anxiety, Depression, and Glutamate System. Front. Nutr. 2021, 8, 744398.
- Herrmann, N.; Chau, S.A.; Kircanski, I.; Lanctôt, K.L. Current and emerging drug treatment options for Alzheimer’s disease: A systematic review. Drugs 2011, 71, 2031–2065.
- Corey-Bloom, J. The ABC of Alzheimer’s disease: Cognitive changes and their management in Alzheimer’s disease and related dementias. Int. Psychogeriatr. 2002, 14, 51–75.
- Haaksma, M.L.; Vilela, L.R.; Marengoni, A.; Calderón-Larrañaga, A.; Leoutsakos, J.S.; Olde Rikkert, M.G.M.; Melis, R.J.F. Comorbidity and progression of late onset Alzheimer’s disease: A systematic review. PLoS ONE 2017, 12, e0177044.
- Pinto, A.; Bonucci, A.; Maggi, E.; Corsi, M.; Businaro, R. Anti-Oxidant and Anti-Inflammatory Activity of Ketogenic Diet: New Perspectives for Neuroprotection in Alzheimer’s Disease. Antioxidants 2018, 7, 63.
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and Oxidative Stress: The Molecular Connectivity between Insulin Resistance, Obesity, and Alzheimer’s Disease. Mediators Inflamm. 2015, 2015, 105828.
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554.
- Ehret, M.J.; Chamberlin, K.W. Current Practices in the Treatment of Alzheimer Disease: Where is the Evidence After the Phase III Trials? Clin. Ther. 2015, 37, 1604–1616.
- Farlow, M.R. Etiology and pathogenesis of Alzheimer’s disease. Am. J. Health Syst. Pharm. 1998, 55, S5–S10.
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031.
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517.
- Ahtiluoto, S.; Polvikoski, T.; Peltonen, M.; Solomon, A.; Tuomilehto, J.; Winblad, B.; Sulkava, R.; Kivipelto, M. Diabetes, Alzheimer disease, and vascular dementia: A population-based neuropathologic study. Neurology 2010, 75, 1195–1202.
- Husain, M.A.; Laurent, B.; Plourde, M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front. Neurosci. 2021, 15, 630502.
- Norton, S.; Matthews, F.E.; Barnes, D.E.; Yaffe, K.; Brayne, C. Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol. 2014, 13, 788–794.
- Rusek, M.; Pluta, R.; Ułamek-Kozioł, M.; Czuczwar, S.J. Ketogenic Diet in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 3892.
- Hersi, M.; Irvine, B.; Gupta, P.; Gomes, J.; Birkett, N.; Krewski, D. Risk factors associated with the onset and progression of Alzheimer’s disease: A systematic review of the evidence. Neurotoxicology 2017, 61, 143–187.
- Cheng, D.; Noble, J.; Tang, M.X.; Schupf, N.; Mayeux, R.; Luchsinger, J.A. Type 2 diabetes and late-onset Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2011, 31, 424–430.
- Chen, L.; Xu, X.; Cao, H.; Li, H. Comparison of effects of different dietary interventions on cognitive function in Alzheimer’s disease: Protocol for systematic review and network meta-analysis. BMJ Open 2021, 11, e042997.
- Lloret, A.; Monllor, P.; Esteve, D.; Cervera-Ferri, A.; Lloret, M.A. Obesity as a Risk Factor for Alzheimer’s Disease: Implication of Leptin and Glutamate. Front. Neurosci. 2019, 13, 508.
- Brown, R.B. Stress, inflammation, depression, and dementia associated with phosphate toxicity. Mol. Biol. Rep. 2020, 47, 9921–9929.
- Paradise, M.; Cooper, C.; Livingston, G. Systematic review of the effect of education on survival in Alzheimer’s disease. Int. Psychogeriatr. 2009, 21, 25–32.
- Caamaño-Isorna, F.; Corral, M.; Montes-Martínez, A.; Takkouche, B. Education and dementia: A meta-analytic study. Neuroepidemiology 2006, 26, 226–232.
- Zhang, T.; Liu, N.; Cao, H.; Wei, W.; Ma, L.; Li, H. Different Doses of Pharmacological Treatments for Mild to Moderate Alzheimer’s Disease: A Bayesian Network Meta-Analysis. Front. Pharmacol. 2020, 11, 778.
- May, B.H.; Feng, M.; Hyde, A.J.; Hügel, H.; Chang, S.Y.; Dong, L.; Guo, X.; Zhang, A.L.; Lu, C.; Xue, C.C. Comparisons between traditional medicines and pharmacotherapies for Alzheimer disease: A systematic review and meta-analysis of cognitive outcomes. Int. J. Geriatr. Psychiatry 2018, 33, 449–458.
- Shearer, J.; Green, C.; Ritchie, C.W.; Zajicek, J.P. Health state values for use in the economic evaluation of treatments for Alzheimer’s disease. Drugs Aging 2012, 29, 31–43.
- Ulamek-Koziol, M.; Pluta, R. To treat or not to treat Alzheimer’s disease by the ketogenic diet? That is the question. Neural Regen. Res. 2020, 15, 857–858.
- Salek, S.S.; Walker, M.D.; Bayer, A.J. A review of quality of life in Alzheimer’s disease. Part 2: Issues in assessing drug effects. Pharmacoeconomics 1998, 14, 613–627.
- Overshott, R.; Byrne, J.; Burns, A. Nonpharmacological and pharmacological interventions for symptoms in Alzheimer’s disease. Expert Rev. Neurother. 2004, 4, 809–821.
- Sandoz, M.; Démonet, J.F.; Fossard, M. Theory of mind and cognitive processes in aging and Alzheimer type dementia: A systematic review. Aging Ment. Health 2014, 18, 815–827.
- Massoud, F.; Léger, G.C. Pharmacological treatment of Alzheimer disease. Can. J. Psychiatry 2011, 56, 579–588.
- Seibert, M.; Mühlbauer, V.; Holbrook, J.; Voigt-Radloff, S.; Brefka, S.; Dallmeier, D.; Denkinger, M.; Schönfeldt-Lecuona, C.; Klöppel, S.; Von Arnim, C.A.F. Efficacy and safety of pharmacotherapy for Alzheimer’s disease and for behavioural and psychological symptoms of dementia in older patients with moderate and severe functional impairments: A systematic review of controlled trials. Alzheimers Res. Ther. 2021, 13, 131.
- Fink, H.A.; Linskens, E.J.; MacDonald, R.; Silverman, P.C.; McCarten, J.R.; Talley, K.M.C.; Forte, M.L.; Desai, P.J.; Nelson, V.A.; Miller, M.A.; et al. Benefits and Harms of Prescription Drugs and Supplements for Treatment of Clinical Alzheimer-Type Dementia. Ann. Intern. Med. 2020, 172, 656–668.
- Hansen, R.A.; Gartlehner, G.; Lohr, K.N.; Kaufer, D.I. Functional outcomes of drug treatment in Alzheimer’s disease: A systematic review and meta-analysis. Drugs Aging 2007, 24, 155–167.
- Mendiola-Precoma, J.; Berumen, L.C.; Padilla, K.; Garcia-Alcocer, G. Therapies for Prevention and Treatment of Alzheimer’s Disease. BioMed Res. Int. 2016, 2016, 2589276.
- Evans, J.G.; Wilcock, G.; Birks, J. Evidence-based pharmacotherapy of Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2004, 7, 351–369.
- Wilcock, G.K.; Ballard, C.G.; Cooper, J.A.; Loft, H. Memantine for agitation/aggression and psychosis in moderately severe to severe Alzheimer’s disease: A pooled analysis of 3 studies. J. Clin. Psychiatry 2008, 69, 341–348.
- Olazarán, J.; Reisberg, B.; Clare, L.; Cruz, I.; Peña-Casanova, J.; Del Ser, T.; Woods, B.; Beck, C.; Auer, S.; Lai, C.; et al. Nonpharmacological therapies in Alzheimer’s disease: A systematic review of efficacy. Dement. Geriatr. Cogn. Disord. 2010, 30, 161–178.
- Ruthirakuhan, M.; Luedke, A.C.; Tam, A.; Goel, A.; Kurji, A.; Garcia, A. Use of physical and intellectual activities and socialization in the management of cognitive decline of aging and in dementia: A review. J. Aging Res. 2012, 2012, 384875.
- Kang, M.J.; Kim, S.M.; Han, S.E.; Bae, J.H.; Yu, W.J.; Park, M.Y.; Ku, S.; Yang, Y. Effect of Paper-Based Cognitive Training in Early Stage of Alzheimer’s Dementia. Dement. Neurocogn. Disord. 2019, 18, 62–68.
- Broom, G.M.; Shaw, I.C.; Rucklidge, J.J. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition 2019, 60, 118–121.
- Gasior, M.; Rogawski, M.A.; Hartman, A.L. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav. Pharmacol. 2006, 17, 431–439.
- Paoli, A.; Rubini, A.; Volek, J.S.; Grimaldi, K.A. Beyond weight loss: A review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur. J. Clin. Nutr. 2013, 67, 789–796.
- Zupec-Kania, B.A.; Spellman, E. An overview of the ketogenic diet for pediatric epilepsy. Nutr. Clin. Pract. 2008, 23, 589–596.
- Norwitz, N.G.; Sethi, S.; Palmer, C.M. Ketogenic diet as a metabolic treatment for mental illness. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 269–274.
- Włodarek, D. Role of Ketogenic Diets in Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019, 11, 169.
- Barry, D.; Ellul, S.; Watters, L.; Lee, D.; Haluska, R., Jr.; White, R. The ketogenic diet in disease and development. Int. J. Dev. Neurosci. 2018, 68, 53–58.
- Park, S.; Zhang, T.; Wu, X.; Qiu, J.Y. Ketone production by ketogenic diet and by intermittent fasting has different effects on the gut microbiota and disease progression in an Alzheimer’s disease rat model. J. Clin. Biochem. Nutr. 2020, 67, 188–198.
- Lange, K.W.; Lange, K.M.; Makulska-Gertruda, E.; Nakamura, Y.; Reissmann, A.; Kanaya, S.; Hauser, J. Ketogenic diets and Alzheimer’s disease. Food Sci. Hum. Wellness 2017, 6, 1–9.
- Boison, D. New insights into the mechanisms of the ketogenic diet. Curr. Opin. Neurol. 2017, 30, 187–192.
- Dhamija, R.; Eckert, S.; Wirrell, E. Ketogenic diet. Can. J. Neurol. Sci. 2013, 40, 158–167.
- Silva, B.; Mantha, O.L.; Schor, J.; Pascual, A.; Plaçais, P.Y.; Pavlowsky, A.; Preat, T. Glia fuel neurons with locally synthesized ketone bodies to sustain memory under starvation. Nat. Metab. 2022, 4, 213–224.
- Higashino-Matsui, Y.; Shirato, K.; Suzuki, Y.; Kawashima, Y.; Someya, Y.; Sato, S.; Shiraishi, A.; Jinde, M.; Matsumoto, A.; Ideno, H.; et al. Age-related effects of fasting on ketone body production during lipolysis in rats. Environ. Health Prev. Med. 2012, 17, 157–163.
- Paoli, A.; Bosco, G.; Camporesi, E.M.; Mangar, D. Ketosis, ketogenic diet and food intake control: A complex relationship. Front. Psychol. 2015, 6, 27.
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of ketone bodies during exercise and training: Physiological basis for exogenous supplementation. J. Physiol. 2017, 595, 2857–2871.
- Qian, M.; Wu, N.; Li, L.; Yu, W.; Ouyang, H.; Liu, X.; He, Y.; Al-Mureish, A. Effect of Elevated Ketone Body on Maternal and Infant Outcome of Pregnant Women with Abnormal Glucose Metabolism During Pregnancy. Diabetes Metab. Syndr. Obes. 2020, 13, 4581–4588.
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52.
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284.
- Wang, Y.H.; Liu, C.L.; Chiu, W.C.; Twu, Y.C.; Liao, Y.J. HMGCS2 Mediates Ketone Production and Regulates the Proliferation and Metastasis of Hepatocellular Carcinoma. Cancers 2019, 11, 1876.
- Hegardt, F.G. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: A control enzyme in ketogenesis. Biochem. J. 1999, 338, 569–582.
- Orii, K.E.; Fukao, T.; Song, X.Q.; Mitchell, G.A.; Kondo, N. Liver-specific silencing of the human gene encoding succinyl-CoA: 3-ketoacid CoA transferase. Tohoku J. Exp. Med. 2008, 215, 227–236.
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102.
- Roubergue, A.; Philibert, B.; Gautier, A.; Kuster, A.; Markowicz, K.; Billette de Villemeur, T.; Vuillaumier-Barrot, S.; Nicole, S.; Roze, E.; Doummar, D. Excellent response to a ketogenic diet in a patient with alternating hemiplegia of childhood. JIMD Rep. 2015, 15, 7–12.
- Kim, J.A.; Yoon, J.R.; Lee, E.J.; Lee, J.S.; Kim, J.T.; Kim, H.D.; Kang, H.C. Efficacy of the classic ketogenic and the modified Atkins diets in refractory childhood epilepsy. Epilepsia 2016, 57, 51–58.
- Zhu, H.; Bi, D.; Zhang, Y.; Kong, C.; Du, J.; Wu, X.; Wei, Q.; Qin, H. Ketogenic diet for human diseases: The underlying mechanisms and potential for clinical implementations. Signal Transduct. Target Ther. 2022, 7, 11.
- Liu, Y.M.; Wang, H.S. Medium-chain triglyceride ketogenic diet, an effective treatment for drug-resistant epilepsy and a comparison with other ketogenic diets. Biomed. J. 2013, 36, 9–15.
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93.
- Croteau, E.; Castellano, C.A.; Richard, M.A.; Fortier, M.; Nugent, S.; Lepage, M.; Duchesne, S.; Whittingstall, K.; Turcotte, É.E.; Bocti, C.; et al. Ketogenic Medium Chain Triglycerides Increase Brain Energy Metabolism in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, 551–561.
- Caprio, M.; Infante, M.; Moriconi, E.; Armani, A.; Fabbri, A.; Mantovani, G.; Mariani, S.; Lubrano, C.; Poggiogalle, E.; Migliaccio, S.; et al. Very-low-calorie ketogenic diet (VLCKD) in the management of metabolic diseases: Systematic review and consensus statement from the Italian Society of Endocrinology (SIE). J. Endocrinol. Investig. 2019, 42, 1365–1386.
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269.
- Greco, T.; Glenn, T.C.; Hovda, D.A.; Prins, M.L. Ketogenic diet decreases oxidative stress and improves mitochondrial respiratory complex activity. J. Cereb. Blood Flow Metab. 2016, 36, 1603–1613.
- Dowis, K.; Banga, S. The Potential Health Benefits of the Ketogenic Diet: A Narrative Review. Nutrients 2021, 13, 1654.
- Gupta, L.; Khandelwal, D.; Kalra, S.; Gupta, P.; Dutta, D.; Aggarwal, S. Ketogenic diet in endocrine disorders: Current perspectives. J. Postgrad. Med. 2017, 63, 242–251.
- Charlot, A.; Zoll, J. Beneficial Effects of the Ketogenic Diet in Metabolic Syndrome: A Systematic Review. Diabetology 2022, 3, 292–309.
- Bostock, E.C.S.; Kirkby, K.C.; Taylor, B.V.; Hawrelak, J.A. Consumer Reports of “Keto Flu” Associated With the Ketogenic Diet. Front. Nutr. 2020, 7, 20.
- Martin, K.; Jackson, C.F.; Levy, R.G.; Cooper, P.N. Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst. Rev. 2016, 9, CD001903.
- Liu, H.; Yang, Y.; Wang, Y.; Tang, H.; Zhang, F.; Zhang, Y.; Zhao, Y. Ketogenic diet for treatment of intractable epilepsy in adults: A meta-analysis of observational studies. Epilepsia Open 2018, 3, 9–17.
- Vanitallie, T.B.; Nonas, C.; Di Rocco, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology 2005, 64, 728–730.
- Palmer, C.M.; Gilbert-Jaramillo, J.; Westman, E.C. The ketogenic diet and remission of psychotic symptoms in schizophrenia: Two case studies. Schizophr. Res. 2019, 208, 439–440.
- Sarnyai, Z.; Kraeuter, A.K.; Palmer, C.M. Ketogenic diet for schizophrenia: Clinical implication. Curr. Opin. Psychiatry 2019, 32, 394–401.
- Murphy, P.; Likhodii, S.; Nylen, K.; Burnham, W.M. The antidepressant properties of the ketogenic diet. Biol. Psychiatry 2004, 56, 981–983.
- Włodarczyk, A.; Cubała, W.J.; Stawicki, M. Ketogenic diet for depression: A potential dietary regimen to maintain euthymia? Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 109, 110257.
- Phelps, J.R.; Siemers, S.V.; El-Mallakh, R.S. The ketogenic diet for type II bipolar disorder. Neurocase 2013, 19, 423–426.
- Kass, H.R.; Winesett, S.P.; Bessone, S.K.; Turner, Z.; Kossoff, E.H. Use of dietary therapies amongst patients with GLUT1 deficiency syndrome. Seizure 2016, 35, 83–87.
- Sullivan, C.R.; Mielnik, C.A.; Funk, A.; O’Donovan, S.M.; Bentea, E.; Pletnikov, M.; Ramsey, A.J.; Wen, Z.; Rowland, L.M.; McCullumsmith, R.E. Measurement of lactate levels in postmortem brain, iPSCs, and animal models of schizophrenia. Sci. Rep. 2019, 9, 5087.
- Rauchenzauner, M.; Klepper, J.; Leiendecker, B.; Luef, G.; Rostasy, K.; Ebenbichler, C. The ketogenic diet in children with Glut1 deficiency syndrome and epilepsy. J. Pediatr. 2008, 153, 716–718.
- Morris, G.; Puri, B.K.; Maes, M.; Olive, L.; Berk, M.; Carvalho, A.F. The role of microglia in neuroprogressive disorders: Mechanisms and possible neurotherapeutic effects of induced ketosis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 99, 109858.
- Castellano, C.A.; Nugent, S.; Paquet, N.; Tremblay, S.; Bocti, C.; Lacombe, G.; Imbeault, H.; Turcotte, É.; Fulop, T.; Cunnane, S.C. Lower brain 18F-fluorodeoxyglucose uptake but normal 11C-acetoacetate metabolism in mild Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 43, 1343–1353.
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530.
- Thelen, M.; Brown-Borg, H.M. Does Diet Have a Role in the Treatment of Alzheimer’s Disease? Front. Aging Neurosci. 2020, 12, 617071.
- Yan, X.; Hu, Y.; Wang, B.; Wang, S.; Zhang, X. Metabolic Dysregulation Contributes to the Progression of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 530219.
- Yao, J.; Chen, S.; Mao, Z.; Cadenas, E.; Brinton, R.D. 2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer’s disease. PLoS ONE 2011, 6, e21788.
- Gough, S.M.; Casella, A.; Ortega, K.J.; Hackam, A.S. Neuroprotection by the Ketogenic Diet: Evidence and Controversies. Front. Nutr. 2021, 8, 782657.
- Maalouf, M.; Rho, J.M.; Mattson, M.P. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res. Rev. 2009, 59, 294–315.
- García-Rodríguez, D.; Giménez-Cassina, A. Ketone Bodies in the Brain Beyond Fuel Metabolism: From Excitability to Gene Expression and Cell Signaling. Front. Mol. Neurosci. 2021, 14, 732120.
- Henderson, S.T. Ketone bodies as a therapeutic for Alzheimer’s disease. Neurotherapeutics 2008, 5, 470–480.
- Brownlow, M.L.; Benner, L.; D’Agostino, D.; Gordon, M.N.; Morgan, D. Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. PLoS ONE 2013, 8, e75713.
- Li, Y.; Sun, H.; Chen, Z.; Xu, H.; Bu, G.; Zheng, H. Implications of GABAergic Neurotransmission in Alzheimer’s Disease. Front. Aging Neurosci. 2016, 8, 31.
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048.
- Whitcomb, D.J.; Hogg, E.L.; Regan, P.; Piers, T.; Narayan, P.; Whitehead, G.; Winters, B.L.; Kim, D.H.; Kim, E.; St George-Hyslop, P.; et al. Intracellular oligomeric amyloid-beta rapidly regulates GluA1 subunit of AMPA receptor in the hippocampus. Sci. Rep. 2015, 5, 10934.
- Kim, D.Y.; Abdelwahab, M.G.; Lee, S.H.; O’Neill, D.; Thompson, R.J.; Duff, H.J.; Sullivan, P.G.; Rho, J.M. Ketones prevent oxidative impairment of hippocampal synaptic integrity through KATP channels. PLoS ONE 2015, 10, e0119316.
- Xu, Y.; Jiang, C.; Wu, J.; Liu, P.; Deng, X.; Zhang, Y.; Peng, B.; Zhu, Y. Ketogenic diet ameliorates cognitive impairment and neuroinflammation in a mouse model of Alzheimer’s disease. CNS Neurosci. Ther. 2022, 28, 580–592.
- Van der Auwera, I.; Wera, S.; Van Leuven, F.; Henderson, S.T. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. 2005, 2, 28.
This entry is offline, you can click here to edit this entry!