1. Adenovirus
Adenovirus (AdV) is a viral agent capable of reaching the CNS. AdV is a member of the
Adenoviridae family. This virus has a linear double-stranded DNA (dsDNA) genome (
Table 1), varying from 25 to 45 Kb [
39]. The viral particle has two main structural elements: the outer capsid and the core [
40]. Within the core, the viral genome is associated with two primary proteins, polypeptide V and polypeptide VII, and a protein called mu (μ), which is rich in arginine [
41]. These proteins function as DNA capacitors within the viral particle [
41]. In addition, the outer capsid presents an icosahedral form, which is constituted by seven proteins (II, III, IIIa, IV, VI, VIII, IX), where II, III, and IV compose the hexon and penton of the viral structure [
42]. On the other hand, researchers have identified other capsid proteins, such as IIIa, VI, VIII, which, together with μ protein, help stabilize the virion capsid [
42]. Finally, also have been reported that VI and μ protein generate infectious viral particles, virus assembly, and viral transcription [
43,
44,
45].
Table 1. Immune evasion and CNS clinical manifestations caused by neurotrophic viruses.
AdV can be found worldwide, and its associated infections can happen at any time of the year. These infections are usually unplanned and are more frequent in children and immunocompromised individuals [
131]. When AdV infects the CNS, it results in neurological disorders such as febrile convulsions, encephalitis, acute disseminated encephalomyelitis, and meningitis (
Table 1) [
48]. AdV serotypes 1 to 7 have been reported in more than 80% of infections in children and adults in countries such as the United States, Canada, the United Kingdom, Taiwan, and South Korea [
131]. In South America, on the other hand, serotype 7 is the most predominant [
131]. Neurological manifestations caused by AdV are rare. It has been observed in pediatric patients that only 1.5% of AdV-infected children have neurological symptoms [
132]. In addition, one study analyzed 48 cases of neurological manifestations caused by AdV in children, where 38% of them presented permanent neurological sequelae [
133]. This virus can infect the CNS, and it can enter through the olfactory nerves, where the virus is transported through the axons of olfactory receptor neurons (ORNs) to the olfactory bulb (
Table 1) [
46]. Virus transport occurs retrogradely, where the virus is internalized at the synapse and then transported by the dynein motor complex into the cell body [
134]. Additionally, AdV can cross the BBB by binding to the coxsackievirus receptor and the adenovirus receptor (CAR) located at the BBB tight junctions [
47]. Thus, when the virus matures, it is released to the basolateral surface and can interact again with these receptors, disrupting the tight junction structures [
47].. In pediatric patients diagnosed with AdV, 3.3% of cases have been found to cause febrile seizures, and there is a 0.4% incidence rate for encephalitis associated with AdV infection. Despite this, AdV is a rare cause of CNS disease, presenting with variable symptoms ranging from mild aseptic meningitis and reversible encephalopathy to severe acute necrotizing encephalopathy (
Figure 1A) [
133].
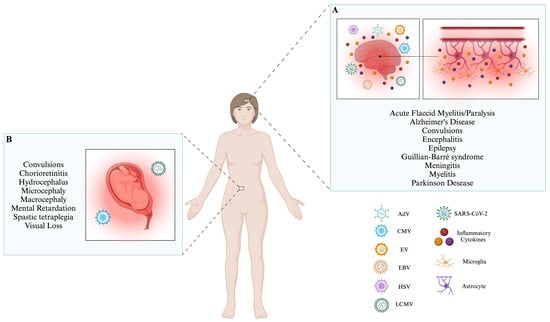
Figure 1. Infection with neurotrophic viruses leads to several conditions that alter the CNS. (A) The neurotrophic viruses mentioned (AdV, CMV, EV, EBV, HSV, LCMV, and SARS-CoV-2) can enter the CNS via potentially different ways; these include via the nasal epithelium, CSF, via hematogenous spread, through nerves, and/or immune cells. When these viruses interact with the CNS, microglia and astrocytes can secrete inflammatory cytokines, including TNF-α, IL-1β, IL-6, and CCL2. Activating this inflammatory process initiates a range of neurological disorders commonly observed across various types of viruses. (B) The presence of LCMV and CMV infections during pregnancy poses significant risks to the proper development of the fetus. Both viruses can cause congenital infections, leading to the improper development of the fetus’s central nervous system. Consequently, the newborn may experience significant long-term effects, such as behavioral or physiological abnormalities (Created by BioRender; License # RU26AQJL7Z).
Infection with AdV activates innate immunity, producing pro-inflammatory and antiviral cytokines such as IFNs [
135,
136]. When the virus infects airway tracheal epithelial cells, it enters through fibers that bind to membrane receptors and adhesion factors [
137]. This effect is enhanced by tumor necrosis factor (TNF)-α and interleukin (IL)-8; this last one also acts as a chemokine [
137]. Both cytokines enhance the expression of the coxsackievirus receptor, CAR, and integrins, facilitating the entry of AdV into the host cells [
137]. At the CNS level,
in vitro experiments have shown that the AdV infection of microglia induces an increase in the expression of inflammatory mediators, especially inducible nitric oxide synthase (iNOS) and TNF-α [
30]. This response is caused by the mitogen-activated protein kinase (MAPK) pathway, where the extracellular signal-regulated kinase (ERK) protein plays a relevant role [
30]. The E1A protein plays a key role in viral infection, modulating transcription and suppressing the host’s innate antiviral responses [
49]. This protein can alter the histone post-translational modifications associated with the latter function, specifically histone H2B [
49]. E1A interacts with the hBre1 complex, preventing the occurrence of H2B monoubiquitination [
49]. This modification is crucial in activating interferon-stimulated genes (ISGs) (
Table 1) [
49]. In addition, it has been noted that the binding of an E1A molecule to two proteins, namely forkhead box family transcription factor (FOXK) and C-terminal binding protein (CtBP), and gene
DCAF7 can effectively inhibit the activation of ISGs [
138]. The virus can also modulate the immune response by changing the active state of the signal transducer and activator of transcription 1 (STAT1). It does this by blocking the dephosphorylation of STAT1, which occurs when the virus cannot interact with TC45 phosphatase. As a result, the virus can control the body’s response to IFNs (
Table 1) [
50]. In addition, AdV possesses proteins encoded in the early transcription region 3 (E3), whose functions can modulate the innate and adaptive immune response (
Table 1) [
51]. One identified protein is E3-gp19K, which can keep major histocompatibility complex (MHC)-I molecules within the endoplasmic reticulum. Furthermore, it hinders the function of the tapsin transporter, which is responsible for processing the peptides presented by MHC-I (
Table 1) [
52]. This mechanism suppresses signals over T cells, destroying cytotoxic T cells [
52].
Clinical studies have extensively highlighted the neurological manifestations of Adenovirus (AdV) infections. This virus can breach the BBB, exploit the olfactory nerve, and infiltrate the CNS. Once inside, AdV utilizes proteins to effectively elude the antiviral immune response, triggering a cascade of clinical manifestations observed in affected individuals.
2. Cytomegalovirus
Another important neurotrophic virus relevant to SLE development is CMV, a member of the
Herpesviridae family, whose genome corresponds to double-stranded DNA (dsDNA) with a size corresponding to 235 Kb (
Table 1) [
53]. The CMV genome can be divided into two main sections: unique long regions and unique short regions [
53]. The virus’s envelope contains glycoproteins that play important roles in virus attachment, entry, maturation, and immune response evasion [
139,
140,
141]. These include glycoproteins such as gB, gH, and UL16 [
54,
140,
142]. Additionally, CMV encodes more than 200 ORFs, encoding 178 proteins. In addition, 32 proteins form the tegument, which plays different roles, from host cell conditioning at the onset of infection to the final stages of virion assembly [
142,
143].
CMV infections are widespread across the globe, particularly in developing nations, where approximately 90% of the population is affected [
144]. CMV infections have a seroprevalence of 90% in the Eastern Mediterranean region and 88% in the Western Pacific and the African region [
144]. Despite this, CMV is a virus that is generally asymptomatic once infection occurs, but in a smaller percentage, immunocompromised people can develop symptoms following infection [
143]. However, CMV infection can trigger alterations at the CNS level, which are infrequent. However, in autopsy cases from immunocompromised patients, 18–28% of them have been affected with CMV-associated neurological manifestations [
145]. CMV can over-regulate the activity of matrix metalloproteases (MMPs) throughout the brain, which can lead to BBB rupture via the degradation of the basement membrane or via the cleavage of the proteins occludin and claudin-5 from tight junctions (
Table 1) [
55]. Also, it is speculated that monocytes may be involved in BBB-associated passage. Once CMV crosses the BBB, it can infect resident cells [
146]. The most prevalent clinical manifestations are polyradiculopathy (inflammation of the spinal nerve roots), myelitis, ventriculitis, dementia, and cranial nerve involvement (
Table 1) [
56,
57]. Additionally, this virus can trigger congenital infection during gestation, directly affecting the CNS or late neurological sequelae such as sensorineural deafness (
Figure 1B) [
147]. The most severe manifestations associated with this phenomenon include microcephaly, periventricular calcifications, convulsions, spastic tetraplegia, and hydrocephalus [
146]. On the other hand, it has been observed that 0.2–2% of newborns develop congenital CMV infection in utero, of which 10–15% have visceral organomegaly, microcephaly with intracranial calcifications, chorioretinitis, jaundice, mental retardation, sensorineural hearing loss (SNHL) and skin lesions [
146]. All these conditions are because CMV can affect various cell populations of the CNS, such as astrocytes, neuronal cells, oligodendrocytes, and microglia [
146]. Additionally, CMV infection can suppress the development of neural stem cells and negatively influence their differentiation (
Figure 1B) (
Table 1) [
58].
During CMV infection, the innate immune response is facilitated by toll-like receptor 2 (TLR2), which identifies the surface glycoproteins gB and gH. This recognition triggers the activation of the nuclear factor kappa B (NF-κB)-dependent signal transduction pathway and the involvement of specificity protein 1 (Sp1) and interferon regulatory factor type 3 (IRF3) [
54,
148,
149,
150]. On the other hand, upon recognizing the presence of the CMV virion, the host’s immune system initiates an innate response by activating IRF7 and interferon-stimulated genes (ISG). This activation leads to the transcription of the ISG54 protein. Additionally, the host releases inflammatory molecules like chemokine ligand (CCL)5, IL-6, and IL-8 [
151,
152,
153].
In vitro analyses have shown that CMV infection in microglia induces the increased secretion of inflammatory cytokines and chemokines, such as TNF-α, IL-6, CCL3, and CCL5 (
Figure 1A) [
154]. Even astrocytes secrete inflammatory mediators in response to CMV infection, including CCL2, IL-8, and CCL3 [
154]. However, CMV can regulate the immune response through the infection of dendritic cells (DCs), which explicitly affects MHC. This event suppresses MHC-I, MHC-II, and co-stimulatory molecules such as CD40 and CD80 (
Table 1) [
59]. In addition, CMV can exert a masking effect through the UL16 glycoprotein to evade the response of natural killer (NK) cells [
141]. Additionally, CMV has been observed to induce phenotypic and functional changes in macrophages, thereby increasing susceptibility to infection (
Table 1) [
60] and causing infected macrophages to lose the ability to present antigens because they lose transcripts and proteins associated with this function, such as the immune evasion genes m152, m4, and m6 (
Table 1) [
61].
While less prone to directly inducing CNS-related pathologies, CMV can rupture the BBB by triggering an overregulation of MMP activity. Nevertheless, its impact extends further as it can significantly affect fetal development, leading to congenital infections that hinder proper neuronal growth. This virus displays a versatile ability to target various cell types, including astrocytes, neuronal cells, oligodendrocytes, and microglia, thus ultimately contributing to the emergence of neurological manifestations. Notably, this virus has been related to the development of autoimmune diseases that affect the CNS, like SLE.
3. Enteroviruses
Other neurotrophic virus that have begun to have a growing incidence and relevance over time are enteroviruses (EVs), members of the
Picornaviridae family. Their genome corresponds to monocistronic single-stranded positive-sense RNA (ssRNA
+), with a length of 7.5 Kb (
Table 1) [
62]. EVs contain four structural proteins called VPs, which are numbered 1 through 4 and are part of the viral capsid [
155]. Additionally, they have non-structural proteins, which promote viral infection and alter the development of the host antiviral response [
63,
156,
157,
158].
EVs can be found on every continent and infect people of all ages [
159]. Geographically, Asia and Europe are the continents most affected by EVs. Worldwide, neurological manifestations associated with these viruses represent 29.4% [
159]. EV infections are usually asymptomatic. However, it has been observed that four human EV species (EV-A to EV-D) are viruses that can trigger CNS-associated clinical conditions [
160]. The EV-A and EV-B enteroviruses are the leading causes of CNS-associated manifestations, among which we find EV-A71, CVB5, and E30, among other species [
159]. EV can enter the CNS through previously infected myeloid cells, which pass through the choroid plexus and penetrate the brain parenchyma (
Table 1) [
35]. These myeloid cells can be infected during extravasation into the choroid plexus and/or passage through the choroid plexus epithelium [
35]. Similarly, EVs can enter the CNS through the BBB, using the endothelial cells that compose this barrier as an entry point and source of viral replication (
Table 1) [
64]. The EV-71 VP1 protein can increase BBB permeability by decreasing the expression of the tight junction protein claudin-5, leading to the induction of BBB leakage [
65]. Furthermore, the virion can invade the brain parenchyma cellularly with elevated BBB leakage through the loosened tight junctions [
65]. Additionally, it has been observed that EV-A71 infects motoneurons, especially at neuromuscular junctions, and thus can invade the CNS (
Table 1) [
66]. Once in the CNS, EVs can affect different cells, such as neural progenitor cells and astrocytes, and neurons have also been shown to be susceptible to EV infection [
161]. This induces aseptic meningitis, encephalitis, and acute flaccid myelitis/paralysis (AFM) development (
Table 1) [
67]. EV-71, which is associated with the CNS, triggers a signaling cascade via the TLR9 receptor that induces the formation of IL-12p40, thus generating a negative effect that induces encephalitis (
Table 1) [
68]. IL-12p40 can damage neurons because it causes excessive neurotoxic nitric oxide (NO) production via iNOS [
68]. The invasion of EV into the CNS can potentially cause cognitive impairments [
69,
70]. In infants as young as three months old, a decline in gross and fine motor skills, problem-solving abilities, socialization, and communication has been observed (
Table 1) [
69]. Similarly, it has been observed that patients who have suffered an EV-71 infection present a delay in neuronal development; in pediatric patients, a reduction in cognitive functioning was observed (
Figure 1A) (
Table 1) [
70]. Notably, EV infection during childhood has been related to attention deficit hyperactivity disorder (ADHD) and epilepsy (
Table 1) [
71]. Furthermore, EV-D68 infection affects both inhibitory (GABAergic) and excitatory (glutaminergic) neurons, leading to a restructuring of the Golgi apparatus at the cellular level. This restructuring results in the formation of a replication organelle [
162]. Additionally, the activity of neurons is reduced upon infection with EV-D68 [
162].
In CNS EV-71 infection, cytokines such as IL-6, IL-8, and CXCL10/IP-10 have been observed in the CSF, which correlates with an increase in the recruitment of monocytes and neutrophils in this fluid [
163]. Additionally, EV-71 infection in primary monocytes generates an immune response mediated by the secretion of IL-1, IL-6, and TGF-α [
164]. In rat neuronal cells cultured
in vitro, it has been observed that, in response to EV infection, both microglia and astrocytes can induce an inflammatory response through the secretion of NO, TNF-α, and IL-1β (
Figure 1A) [
165]. The absence of antiviral responses associated with type I interferon (IFN-I) may be because of the presence of the virus’s non-structural proteins in processes that enable the secretion of these cytokines (
Table 1). The non-structural protein A2 of EV can perform this effect by suppressing the IFN-β gene [
158]. Another mechanism described for strain EV-71 involves the suppression of the antiviral response, mediated by the non-structural protein 3C, which negatively regulates the cytosolic receptor retinoid acid-inducible gene I (RIG-I) pathway (
Table 1) [
63].
Over time, enteroviruses have gained increasing prominence worldwide due to their significant role in the development of neurological manifestations. These viruses can breach the BBB by utilizing infected immune cells or motor neurons as a gateway into the CNS. Upon entry, they provoke an inflammatory response and cause cellular damage, resulting in distinct clinical manifestations associated with these infections. The development of autoimmune diseases like SLE and the possible link with this viral infection are poorly described.
4. Epstein-Barr Virus
Like CMV, Epstein–Barr Virus (EBV) is another
Herpesviridae family member with great neurotrophic potential and is relevant in developing autoimmune diseases, especially SLE. The EBV genome corresponds to double-stranded DNA (dsDNA) with a length of around 172 Kb (
Table 1) [
75]. This virus contains seven structural proteins, which have fundamental roles for the virus; for example, BFRF3 is associated with the assembly of the viral capsid [
166,
167]. Several glycoproteins have diverse functions, such as the fusion of the viral envelope on the host cell, viral propagation, and the disruption of the antiviral response [
168,
169,
170,
171]. Additionally, this virus presents tegument proteins, whose functions are varied. They are associated with the release of mature virions, infectivity, viral production, and the inhibition of the host’s antiviral response [
172,
173,
174,
175,
176].
It has been observed that EBV infections are not selective based on age, as they have the potential to impact individuals of all age groups, including both children and adults [
177,
178]. In China, it has been observed in recent years that in children who have suffered EBV infections, approximately 0.6% of them present neurological manifestations [
179]. EBV infection can modulate the BBB because this virus infects microvascular endothelial cells. This induces inflammatory and endothelial markers that can trigger potential neurological manifestations (
Table 1) [
76]. Upon infection, the EBV dUTPase protein induces the expression of pro-inflammatory cytokines such as IL-6 and IL-1β in microvascular endothelial cells and microglia, as well as TNF-a in astrocytes [
77]. These cytokines plus IFN-γ alter the integrity of the BBB by regulating the expression of genes encoding for proteins that are involved in forming and maintaining the tight junctions between endothelial cells in the capillaries of the BBB, as well as modulating cell adhesion and the extracellular matrix [
77]. EBV can affect critical CNS cells, such as neurons and glial cells [
180,
181]. EBV can trigger encephalitis, in whose pathology CD4
+ T and CD8
+ T cells play an essential role by generating immune checkpoint inhibitors [
9,
182]. Other pathologies associated with the CNS induced by EBV are acute transverse myelitis, cerebellitis, ataxia, and Guillian–Barré syndrome (
Figure 1A) (
Table 1) [
78,
79]. When caused by EBV infection, cerebellar ataxia involves the presence of self-reactive antibodies (referred to as anti-neuronal antibodies) [
183]. These antibodies are produced due to a phenomenon known as mimetism, where similarities between EBV proteins and neuronal antigens trigger their production [
183]. On the other hand, EBV can trigger neurodegenerative diseases, including Alzheimer’s Disease (AD) and Parkinson’s disease (
Table 1) [
80,
81]. An
in silico analysis determined that EBV infection is associated with AD development due to the formation of peptide aggregates, specifically a 12-amino acid peptide derived from the glycoprotein gM (
Figure 1A) [
80]. Additionally, it has been observed that neuronal cells exposed to EBV and gM induce an environment that promotes neuroinflammation due to the positive upregulation of specific inflammatory cytokines such as IL-1β, IL-6, and TNF-α [
31]. On the other hand, Parkinson’s disease has also been associated with EBV infection because it is caused by a molecular mimetism between the C-terminal region of α-synuclein and a repeat region in the latent membrane protein 1 (LMP1) protein from EBV [
81].
It has been reported that EBV-infected patients show elevated levels of IL-6, IL-18, IFN-γ, TNF-α, and IL-10 in their serum [
184]. At the CNS level, it has been observed in glial cell lines that, upon stimulation with EBV, these induce an increase in the expression of inflammatory cytokines such as IL-6 and IL-1β (
Figure 1A) [
180]. EBV possesses some mechanisms to evade the host antiviral response through different proteins. BILF1 inhibits NLRP3 inflammasome activation and subsequent pyroptosis, which inhibits viral replication (
Table 1) [
82]. This is because an interaction occurs between the BILF1 protein and the host UFL1 ligase, which generates UFMylations on mitochondrial antiviral signaling (MAVS), causing it to pack into vesicles (
Table 1) [
82]. On the other hand, EBV also induces an evasion pathway for IFN-I secretion (
Table 1) [
83]. Tyk2 phosphorylation is inhibited by cytoplasmic envelopment protein 2 (BGLF2), causing STAT2 and STAT3 not to be activated, resulting in the non-expression of IFN-I-associated genes [
83]. The tegument protein, BFRF1, acts on IKKi, inhibiting its kinase activity; this blocks IRF3 translocation to the nucleus and inhibits the IFN-I pathway, specifically IFN-β (
Table 1) [
84]. Another tegument protein, BPLF1, has a deubiquitinase activity, which removes TBK1 ubiquitins while suppressing IRF3 dimerization [
185]. Additionally, BKRF4 has immune response inhibitory activity since this protein interacts with histones H2A-H2B, causing EBV to have a mimetic effect since the host cannot detect the signals in response to DNA damage [
173]. Also, the EBV-encoded miR-BART16 can suppress the production of IFN-stimulated genes and hinder the anti-proliferative impact of IFN-α on B cells during latent infection (
Table 1) [
186]. On the other hand, it is known that EBV can infect monocytes through a gp25–gp42–gp85 complex, which interacts with the membrane of this immune cell, triggering apoptosis and inhibiting the development of DCs (
Table 1) [
85]. Likewise, it has been observed that the glycoprotein gp42 can interact with human leukocyte antigen (HLA)-DR1 of MHC-II, promoting B cell infection [
187]. EBV infection over B cells can generate reprogramming on them, decreasing CXCR4 expression, modulating CD23 expression, and even regulating more than 11,000 genes associated either metabolically or phenotypically (
Table 1) [
86,
87,
88].
The relevance of EBV is due to its modulatory effect on the cells of the immune system. This virus can enter the CNS through the BBB and then interact with neurons and glial cells, triggering an inflammatory and tissue-damaging process that leads to CNS pathologies. Notably, the role of EBV in autoimmune diseases such as MS and SLE makes the timely diagnosis and development of new treatments to prevent these diseases necessary.
5. Herpes Simplex Virus
HSV infections are responsible for most genital and neonatal infections globally, accounting for approximately 85% of cases [
189]. Additionally, there has been a rise in the age at which individuals first engage in sexual activity [
189]. HSV infections are most prevalent in Africa and America [
190]. HSV can affect the CNS via local spread from the periphery or viremia. In the case of HSV-1 infections, there are believed to be three routes by which it can infect the CNS [
92]. The first is from the primary oropharyngeal infection site, which can reach the brain via the trigeminal or olfactory nerves. The second involves the same neural pathways but is caused by a reactivation of initial infection in the periphery and, finally, the reactivation of latent HSV-1 in situ in the brain (
Table 1) [
92]. The trigeminal and olfactory nerve pathways allow HSV to infect the CNS by circumventing cellular barriers, such as the blood–brain and cerebrospinal fluid barriers, leading to infection (
Table 1) [
93].
On the other hand, HSV-1 infection triggers Golgi stress through changes in morphology and function. The GM130 protein is downregulated, and there is an increase in apoptotic cells, regulating the stress response of the Golgi apparatus. This contributes to alterations in the structure and function of the BBB by downregulating occludin and claudin-5 [
94]. In addition, Golgi apparatus fragmentation after infection leads to increased Ca
2+ release, stimulating endothelial NOS (eNOS) activation and modulating the NO and vascular endothelial growth factor (VEGF) effectors that lead to BBB rupture [
95]. When HSV infects the CNS, several cells are affected, mainly neurons, but astrocytes and oligodendrocytes have also been susceptible to HSV infection [
191]. Despite this, the exact mechanism by which HSV-1 can destroy neuronal cells is unknown. However, this could be due to direct injury caused by the virus or immune-mediated cell injury [
93]. HSV can induce meningitis and acute encephalitis, affecting individuals of all age ranges (
Figure 1A) (
Table 1) [
10,
11,
96]. Encephalitis caused by HSV is uncommon since it has been observed to have an annual incidence rate estimated at 1 per 250,000–500,000 inhabitants [
192]. The principal causative agent is HSV-1 [
192]. Additionally, it has been observed that HSV-1 is present in 2.8% of Korean patients with encephalitis [
193]. On the other hand, HSV-2 is found in 5.7% of patients with aseptic meningitis [
193]. Encephalitis caused by HSV-2 affects glial cells, causing hemorrhagic necrosis in the temporal lobe [
96]. Studies suggest that the epinephrine and corticosterone receptors expressed on sensory and sympathetic neurons modulate the potential for HSV infection [
96]. Furthermore, the infection of the CNS with HSV-1 can result in cognitive impairment, although it does not reach the severity of dementia or other neurodegenerative conditions such as AD (
Table 1) [
97].
HSV infection triggers an immune response associated with pathogen-associated molecular patterns (PAMPs) through the TLR pathway, activating different proteins; among these are RIG-1, melanoma differentiation-associated protein 5 (MDA5), cyclic GMP-AMP synthase (cGAS), interferon-inducible protein AIM2 (AIM2), interferon-gamma-inducible protein 16 (IFI16), and DEAH-box proteins (DHX) [
194,
195,
196]. This pathway initiates intracellular signaling pathways that trigger the production and release of molecules that hinder viral replication [
194,
195,
196]. Additionally, the presence of TLR2 in antigen-presenting cells (APCs) activates NF-κB, which induce inflammatory cytokines production such as TNF, IL-1, IL-6, IL-8, IFN-γ, CXCL5, CXCL9, CXCL10, and CCL3 [
197,
198,
199,
200]. Microglia play a fundamental role in combating HSV infection in the CNS because they induce antiviral responses mediated by cGAS-STING, triggering IFN-I expression [
201]. In astrocytes, infection with HSV-1 induces this cell population to secrete CXCL1, which causes the increased migration of neutrophils to the site of infection [
202].
Like other viruses, HSV can modulate the host’s antiviral response. HSV-2 inhibits the ability of the cells it infects to recognize viral dsRNA, causing changes in the IFN-I pathways (
Table 1) [
98]. The early viral protein ICP0 can also inhibit IRF3, blocking the transcription of target genes associated with this factor (
Table 1) [
99]. On the other hand, to prevent the activation of protein kinase R (PKR), HSV-1 utilizes the late genes γ34.5 and Us11 to hinder the activation of this protein. As a result, the translation of eIF2A remains inactive, leading to the inability to suppress the translation of viral mRNAs [
203]. The glycoproteins from EBV can also modulate the immune response, where gC binds to the C3b component of the complement and prevents the interaction of C5 and properdin with C3b. As a result, it hinders the activation of both the classical and alternative pathways of complement activation (
Table 1) [
91]. Moreover, HSV can regulate the adaptative immune responses. The glycoprotein gE hinders the ability of IgG antibodies to carry out their intended function by attaching to the Fc section (
Table 1) [
90]. HSV-2 can impact DCs, leading to their apoptosis and hindering the activation of T cells (
Table 1) [
100]. Furthermore, it has been observed that HSV-1 utilizes the γ34.5 protein to hinder autophagosome development within infected cells. As a result, the ability to present antigens to T cells is impeded, disrupting their regulation [
204]. The effect associated with the antigens present is HSV is accomplished using a VHS-RNase. This enzyme facilitates the breakdown of host mRNA, decreasing the production of antiviral components [
101]. The combination of these factors, along with the presence of the ICP47 protein, leads to a reduction in the expression of MHC-I on the surface of cells and a decrease in MHC-II levels. This impairment affects the ability to present antigens and ultimately hinders cellular and humoral immune responses (
Table 1) [
101,
102,
103,
104,
205].
Since HSV infection is one of the most common worldwide, the ability of this virus to trigger CNS-associated manifestations is concerning. HSV can use the trigeminal and olfactory nerves to invade the CNS and, by stressing the Golgi apparatus, can modulate the BBB’s structure. By targeting cells such as neurons, astrocytes, and oligodendrocytes, this virus provokes neurological pathologies. Recent findings associate HSV viral infection with autoimmune disease, but little is known in this field, and more research is needed.
6. Lymphocytic Choriomeningitis Virus
In contrast to the mentioned viruses belonging to the
Herpesviridae family, the LCMV virus belongs to the
Arenaviridae family, which is characterized by being an enveloped virus with a bi-segmented negative-sense single-stranded RNA (ssRNA
−) genome (
Table 1) [
105], which corresponds to a short segment (S) of 3.5 Kb and a long segment (L) of 7.2 Kb [
105]. The RNA S section encodes for highly relevant structural proteins, specifically nucleoproteins and glycoproteins [
206]. On the other hand, the RNA L section codes for the L protein corresponding to the RNA polymerase of the virus. The N-terminal region acts as an RNA endonuclease [
207,
208,
209]. Additionally, this genomic segment codes for a protein containing a zinc fingers sequence, which may be associated with RNA binding with a regulatory function [
210]. The LCMV nucleoprotein (NP) is involved in processes that facilitate the intracellular spread of the virus, transcription, translation, and genome replication. Furthermore, it acts on host immune responses [
211,
212,
213,
214,
215]. LCMV glycoproteins are associated with receptor-binding infection persistence and are related to virus fusion [
216,
217].
The primary reservoir for LCMV is rodents, particularly mice [
218]. Viral human infection is due to direct contact with infected animals [
218]. Over the years, this virus has been found in several countries worldwide because LCMV is prevalent in American, African, Asian, and European mice [
219]. Studies performed in the last decade in Iraq and Finland have indicated that 5.1% and 5%, respectively, of people who have been affected by a neuroinvasive disease are positive for LCMV [
219]. LCMV can infect the ependyma, choroid plexus, and brain meningeal regions. All this leads to LCMV infection generating BBB alteration (
Table 1) [
106]. CD8
+ T cells are relevant to combat LCMV infection. However, this lymphocyte population can modulate the BBB through perforin secretion or by secreting chemokines that recruit monocytes and neutrophils, inducing tissue damage [
107,
108,
109]. In a murine model, especially in rats, it has been observed that astrocytes and Bergmann glial cells are the main brain parenchymal cells infected by LCMV. The virus can spread through these cells, specifically neurons in the cerebellum, olfactory bulb, dentate gyrus, and periventricular region [
220]. Individuals infected with LCMV often have CNS-associated problems, such as aseptic meningitis (
Table 1) [
10]. The symptoms associated with developing LCMV infection are varied and may include fever, myalgia, headache, and malaise [
221]. However, some patients develop photophobia, vomiting, and nuchal rigidity [
221]. Once LCMV infection of the CNS occurs, cells such as neurons and astrocytes are affected [
222]. On the other hand, when a woman acquires LCMV infection during the second trimester of pregnancy, it has been observed that this infection can lead to the development of congenital disabilities in the fetus [
110,
111]. These defects may include hydrocephalus and chorioretinitis (
Figure 1B) (
Table 1) [
110]. Also, when pregnant women contract LCMV, their babies can develop conditions such as macrocephaly, microcephaly, chorioretinopathy, and in some cases, experience severe neurological effects like spastic quadriparesis, seizures, visual loss, or mental retardation (
Figure 1A) (
Table 1) [
112]. These congenital effects are due mainly to the fact that LCMV infection affects neuroblasts, which are calcified in the brain’s periventricular region by congenital infection [
223]. In addition, LCMV alters neuron migration [
223]. Also, murine models, particularly rats, have demonstrated the occurrence of neurodegenerative manifestations (
Table 1) [
224]. LCMV-infected rats have a movement of T cells toward the brain [
224]. This migration serves to control glial infection while also contributing to brain degeneration. Notably, this degeneration does not affect the hippocampus, an area of the brain highly associated with severe neuropsychiatric diseases [
202].
The activation of the innate immune response against LCMV infection is linked to the TLR2 and MyD88 pathways [
225]. These pathways are responsible for triggering the IFN-I production, particularly IFN-α [
225]. At the same time, MyD88 is associated with activating antiviral CD8
+ T cells to take antiviral action against LCMV [
225]. The secretion of IL-27 by B cells is regarded as having antiviral properties, as it facilitates the survival of the CD4
+ T cells that target LCMV and encourages antibody class switching [
226]. In a study conducted on mice, it was demonstrated that there is a distinct immune response to LCMV infection in males and females [
227]. The CSF from males has lower levels of CCL5, IL-1α, and IL-1β compared to females and has a lower activation of APCs [
227]. By the eighth day after infection, males exhibit decreased levels of IFN-γ compared to females. During this stage of viral infection, the production of this cytokine relies on T cells. Therefore, this observation suggests a potential disruption in the functioning of T cells in males [
227]. The relevance of IFN-γ in the CD4
+ T response against LCMV in the CNS is significant, as this cytokine plays a crucial role in inducing MHC-II expression [
189]. Even DCs, in response to infection, can secrete inflammatory cytokines, IL-12 and IL-23, through TLR activation [
228]. In the CNS, in response to LCMV infection, both microglia and astrocytes can produce inflammation-associated molecules such as TNF-α, CCL5, and CCL2 (
Figure 1A) [
229]. Additionally, glial cells recognize LCMV through TLR2, thus activating the MyD88 pathway [
229]. However, LCMV has mechanisms to evade the antiviral response. The NP protein plays a crucial role in suppressing the production of IFN (
Table 1). Its exoribonuclease activity removes viral RNA, prevents phosphorylation IRF3, and does not migrate to the nucleus, suppressing IFN-I production [
113]. Additionally, the NP protein can interact with the kinase domain of the IKKε protein, preventing it from phosphorylating the IRF-3 protein. Consequently, this interaction hampers the production of IFN-I (
Table 1) [
114].
LCMV infection has shown its ability to trigger CNS-associated pathologies, where CD8+ T cells play a crucial role in modulating the BBB. However, this virus not only affects children and adults but can also affect fetuses through congenital infection, principally affecting neurons and interfering with the correct neuronal development, generating severe and irreversible pathologies. The relationship between the infection of LCMV and its role in autoimmune diseases like SLE is still understood.
7. Severe Acute Respiratory Syndrome Coronavirus-2
Unlike all the neurotrophic viruses mentioned in this section, SARS-CoV-2 has significantly impacted health worldwide in recent years and is mainly characterized by the respiratory disease it triggers. The recently described SARS-CoV-2 is a member of the
Coronaviridae family, which is characterized by a single-stranded RNA genome that is positive-sense (ssRNA
+) and non-segmented (
Table 1). The size of this genome can fluctuate between 27 and 32 Kb [
115,
116]. In contrast, the length of SARS-CoV-2 specifically falls within the range of 29.8 to 29.9 Kb [
230]. This virus encodes for four structural proteins: spike protein (S), envelop protein (E), membrane protein (M), and nucleocapsid protein (N) [
231,
232,
233]. Additionally, the non-structural proteins (NSPs) that present these proteins play crucial roles in virus pathogenicity, genome replication, and the modulation of the immune response [
117,
234,
235]. Finally, accessory proteins are relevant in evading the immune response [
118,
119,
236,
237,
238,
239].
The SARS-CoV-2 virus caused a global health crisis, where no country or continent was exempt from suffering its effects. It has been observed that socioeconomic status is a factor that increases the incidence of this virus; in sectors that have a lower socioeconomic status, there is a higher SARS-CoV-2 positivity rate compared to sectors that have a better status [
240]. On the other hand, SARS-CoV-2-hospitalized patients who previously did not have a neurological disease frequently had CNS-associated manifestations (approximately 33%) [
241]. This virus can enter the CNS because it can cross the BBB by altering the tight junctions (
Table 1) [
120]. This occurs because the virus can modulate the expression of MMP9, an endopeptidase necessary for remodeling the extracellular matrix, causing collagen IV degradation [
120]. Also, it has been observed that components of the spike protein, such as S1 and S2, cause BBB leakage induction [
121]. Additionally, SARS-CoV-2 can interact with cranial nerves IX (glossopharyngeal nerve) and X (vagal nerve) in such a way that it can potentially enter the CNS via this pathway; this is because the ACE-2, NRP1, and TMPRSS2 proteins are found in these cranial pairs (
Table 1) [
122].
Astrocytes are the primary target of SARS-CoV-2 infection in the CNS, as the virus utilizes the NRP1 receptor to enter these cells and establish itself as a site for viral replication [
32]. Astrocytes infected by this virus have decreased glutamine metabolism intermediates (glutamate and GABA) [
242]. In addition to NRP1, astrocytes possess other molecules that allow the SARS-CoV-2 virus to enter the target cells. These additional molecules include TMPRSS-2 and ACE-2 proteins [
32]. The presence of the virus in the CNS causes neuronal symptoms by inducing neuron–neuron or neuron–glia fusion [
243]. The process of neuronal fusion occurs sequentially and, after that, significantly impairs neuronal function [
243]. Once SARS-CoV-2 interacts with the CNS, it can generate pathologies such as encephalitis and encephalopathy, seizure and epilepsy, and even neurodegenerative diseases, such as AD (
Figure 1 A) (
Table 1) [
123,
124,
125,
126]. It has been noted that SARS-CoV-2 has the potential to contribute to the development of AD. This observation is supported by the fact that viral infection causes modifications in gene expression, particularly in genes linked to the Wnt pathway, which is known to be disrupted in this neurodegenerative condition [
123]. There are additional pathways that could contribute to the progression of this disease. These pathways include changes in inflammation, the processing of amyloid proteins, cellular trafficking linked to the transport of proteins in the endoplasmic reticulum, the breakdown of proteins involved in ubiquitin, and the regulation of calcium levels [
123].
It is widely recognized that SARS-CoV-2 induces a cytokine storm, leading to an increase in inflammatory cytokines such as IL-8, IL-15, IFN-α2a, IFN-γ, CXCL10, CCL2, and TNF-α [
244,
245]. Regarding transcription, there is an increase in the activation of inflammatory pathways associated with NF-κB and IL-6-STAT3 [
246]. Simultaneously, the E protein of SARS-CoV-2 interacts with TLR2, initiating a signaling pathway that involves NF-kB. This activation enables the transcription of inflammatory cytokines and chemokines, such as CXCL8, which share a similar context [
247]. Likewise, the ORF7b protein promotes IFN-I signaling and the expression of inflammatory cytokines such as TNF-α and IL-6 [
238]. In individuals with CNS infection caused by SARS-CoV-2, inflammatory cytokines like IL-4, IL-6, and IL-12 are released by microglia and astrocytes (
Figure 1A) [
125]. On the other hand, SARS-CoV-2 can modulate the host’s immune response and evade antiviral responses. The N protein affects RIG-I recognition on dsRNA (
Table 1) [
127]. Additionally, it has been observed that both the N protein and NSP5 protein can attenuate the formation of antiviral granules and suppress the IFN pathway (
Table 1) [
127,
128]. The mechanism of inhibition in this pathway is related to the inhibition of TBK1 and IRF3 phosphorylation, resulting in the impossibility of IRF3 translocating to the nucleus [
127]. The non-structural protein NSP5 can cleave the essential modulator NF-κB (NEMO), a relevant kinase for the RIG-I pathway (
Table 1) [
129]. Another non-structural protein, NSP12, affects the response to IFN by preventing the nuclear translocation of IRF3 (
Table 1) [
117]. The C-terminal segment of the ORF3b protein from the SARS-CoV-2 virus can reduce the activity of IFN-I by inhibiting the movement of IRF3 to the nucleus [
236]. The binding of ORF6 to STAT1 prevents phosphorylated STAT1 from migrating to the nucleus, inhibiting antiviral responses and increasing SARS-CoV-2 viral replication [
237]. On the other hand, ORF10 can act on the signaling pathway associated with MAVS because this accessory protein can induce MAVS autophagy, thus inhibiting the IFN-I response and promoting viral replication simultaneously (
Table 1) [
119]. This virus can also module the immune chemotaxis because the N protein can bind to these molecules, including CXCL12B, CCL8, CCL26, and CXCL10 (
Table 1) [
130]. Additionally, ORF7a generates a molecular mimetic of β2-microglobulin (β2m) that can interact with MHC-I, inducing the antigen presentation processes and, consequently, the antiviral response (
Table 1) [
118].
In contrast to the significant respiratory impact caused by SARS-CoV-2 globally, there has been a recent recognition of its relevance in developing neurological diseases. These conditions arise from the virus’s invasion of the CNS through the BBB and cranial nerves. Simultaneously, this invasion triggers an evasion of the antiviral immune response, leading to substantial damage to neuronal cells. The role of this virus in developing autoimmune diseases remain to be elucidated.
Each virus described is characterized and associated mainly with pathologies not directly linked to the CNS. However, each of these viruses can invade the CNS in different ways, where two mechanisms are the most relevant for their infection; these include BBB modulation and entry through nerves [
48,
55,
65,
92,
121,
122]. These viruses, once inside this system, can interact and infect various cells, such as astrocytes, which are one of the most abundant cell types in the brain; they play a neuronal support role and can participate in synapses [
161,
180,
181,
220,
242,
243]. At the same time, this infection process leads to the development of an innate response by immune cells specific to this tissue, such as microglia, and a subsequent adaptive response [
30,
54,
125,
154,
165,
180,
201,
225]. All of this is to eliminate the virus. However, each virus can evade the host’s immune response through its different proteins. This leads principally to the virus not being eliminated and the generation of neurological clinical pictures and even neurodegenerative diseases [
48,
50,
56,
57,
59,
67,
71,
72,
80,
81,
84,
96,
99,
110,
113,
119,
123].
This entry is adapted from the peer-reviewed paper 10.3390/brainsci14010059